
Key Points
-
Congenital diarrhoeal disorders (CDDs) are a group of rare inherited enteropathies with a typical onset early in the life
-
These disorders are challenging clinical conditions because of the severity of the clinical picture and the broad range of diseases in differential diagnosis
-
To simplify the approach to these conditions, a classification in four groups according to the main pathogenetic mechanism has been proposed
-
The number of conditions included within the CDDs group has gradually increased, and many new genes have been identified, opening new diagnostic and therapeutic perspectives
-
Clinically actionable molecular methods to diagnosis CDDs, and other monogenic disorders, have markedly improved in recent years
-
Continued research is focused on identifying novel CDDs, and to define in detail the pathogenesis of established disorders that might provide novel therapeutic options to ameliorate morbidity and mortality
Abstract
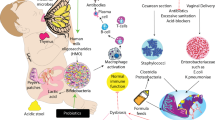
Congenital diarrhoeal disorders (CDDs) represent an evolving web of rare chronic enteropathies, with a typical onset early in life. In many of these conditions, severe chronic diarrhoea represents the primary clinical manifestation, whereas in others diarrhoea is only a component of a more complex multi-organ or systemic disorder. Typically, within the first days of life, diarrhoea leads to a life-threatening condition highlighted by severe dehydration and serum electrolyte abnormalities. Thus, in the vast majority of cases appropriate therapy must be started immediately to prevent dehydration and long-term, sometimes severe, complications. The number of well-characterized disorders attributed to CDDs has gradually increased over the past several years, and many new genes have been identified and functionally related to CDDs, opening new diagnostic and therapeutic perspectives. Molecular analysis has changed the diagnostic scenario in CDDs, and led to a reduction in invasive and expensive procedures. Major advances have been made in terms of pathogenesis, enabling a better understanding not only of these rare conditions but also of more common diseases mechanisms.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others


References
Berni Canani, R., Terrin, G. Recent progress in congenital diarrheal disorders. Curr. Gastroenterol. Rep. 13, 257–264 (2011).
Abou Ziki, M. D. & Verjee, M. A. Rare mutation in the SLC26A3 transporter causes life-long diarrhoea with metabolic alkalosis. BMJ Case Rep. bcr2014206849 (2015).
Passariello, A. et al. Diarrhea in neonatal intensive care unit. World J. Gastroenterol. 16, 2664–2668 (2010).
Pezzella, V. et al. Investigation of chronic diarrhoea in infancy. Early Hum. Dev. 89, 893–897 (2013).
Wiegerinck, CL. et al. Loss of syntaxin 3 causes variant microvillus inclusion disease. Gastroenterology 147, 65–68 (2014).
Xin, B. & Wang, H. Multiple sequence variations in SLC5A1 gene are associated with glucose-galactose malabsorption in a large cohort of Old Order Amish. Clin. Genet. 79, 86–91 (2011).
Berni Canani, R., Terrin, G., Cardillo, G., Tomaiuolo, R. & Castaldo, G. Congenital diarrheal disorders: improved understanding of gene defects is leading to advances in intestinal physiology and clinical management. J. Pediatr. Gastroenterol. Nutr. 50, 360–366 (2010).
Terrin, G. et al. Congenital diarrheal disorders: an updated diagnostic approach. Int. J. Mol. Sci. 13, 4168–4185 (2012).
Fiskerstrand, T. et al. Familial diarrhea syndrome caused by an activating GUCY2C mutation. N. Engl. J. Med. 366, 1586–1595 (2012).
Basu, N., Arshad, N. & Visweswariah, S. S. Receptor guanylyl cyclase C (GC-C): regulation and signal transduction. Mol. Cell. Biochem. 334, 67–80 (2010).
Li, C. & Naren, A. P. CFTR chloride channel in the apical compartments: spatiotemporal coupling to its interacting partners. Integr. Biol. (Camb.) 2, 161–177 (2010).
Haas, J. T. et al. DGAT1 mutation is linked to a congenital diarrheal disorder. J. Clin. Invest. 122, 4680–4684 (2012).
Halac, U. et al. Microvillous inclusion disease: how to improve the prognosis of a severe congenital enterocyte disorder. J. Pediatr. Gastroenterol. Nutr. 52, 460–465 (2011).
van der Velde, K. J. et al. An overview and online registry of microvillus inclusion disease patients and their MYO5B mutations. Hum. Mutat. 34, 1597–1605 (2013).
Schnell, U. et al. Absence of cell-surface EpCAM in congenital tufting enteropathy. Hum. Mol. Genet. 22, 2566–2571 (2013).
Fabre, A. et al. Syndromic (phenotypic) diarrhoea of infancy/tricho-hepato-enteric syndrome. Arch. Dis. Child. 99, 35–38 (2014).
Knowles, B. C. et al. Myosin Vb uncoupling from RAB8A and RAB11A elicits microvillus inclusion disease. J. Clin. Invest. 124, 2947–2962 (2014).
Ruemmele, F. M. et al. Loss-of-function of MYO5B is the main cause of microvillus inclusion disease: 15 novel mutations and a CaCo-2 RNAi cell model. Hum. Mutat. 31, 544–551 (2010).
Thoeni, C. & Cutz, E. Pediatric and perinatal pathology: SY21–23 recent advances in molecular pathology of microvillous inclusion disease (MVID). Pathology 46 (Suppl. 2), S34 (2014).
Kravtsov, D. et al. Myosin 5b loss of function leads to defects in polarized signalling: implication for microvillus inclusion disease pathogenesis and treatment. Am. J. Physiol. Gastrointest. Liver Physiol. 307, G992–G1001 (2014).
Rodriguez, O. C. & Cheney, R. E. Human myosin-Vc is a novel class V myosin expressed in epithelial cells. J. Cell Sci. 115, 991–1004 (2002).
Girard, M. et al. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillus inclusion disease. Hepatology 60, 301–310 (2014).
Stephensky, P. et al. Persistent defective membrane trafficking in epithelial cells of patients with familial hemophagocyticlymphohistiocytosis type 5 due to STXBP2/MUNC18–12 mutations. Pediatr. Blood Cancer 60, 1215–1222 (2013).
Kozan, P. A. et al. Mutation of EpCAM leads to intestinal barrier and ion transport dysfunction. J. Mol Med. (Berl.) http://dx.doi.org/10.1007/s00109-014-1239-x.
Salomon, J. et al. Genetic characterization of congenital tufting enteropathy: epcam associated phenotype and involvement of SPINT2 in the syndromic form. Hum. Genet. 133, 299–310 (2014).
Fabre, A. et al. Novel mutations in TTC37 associated with tricho-hepato-enteric syndrome. Hum. Mutat. 32, 277–281 (2011).
Fabre, A. et al. SKIV2L mutations cause syndromic diarrhea, or trichohepatoenteric syndrome. Am. J. Hum. Genet. 90, 689–692 (2012).
Smith, S. B. et al. Rfx6 directs islet formation and insulin production in mice and humans. Nature 463, 775–780 (2010).
Du, A. et al. Arx is required for normal enteroendocrine cell development in mice and humans. Dev. Biol. 365, 175–188 (2012).
Zhu, X. et al. Disruption of PC1/3 expression in mice causes dwarfism and multiple neuroendocrine peptide processing defects. Proc. Natl Acad. Sci. USA 99, 10293–10298 (2002).
Gradwohl, G., Dierich, A., LeMeur, M. & Guillemot, F. Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl Acad. Sci. USA 97, 1607–1611 (2000).
Wang, J. et al. Mutant neurogenin-3 in congenital malabsorptive diarrhea. N. Engl. J. Med. 355, 270–280 (2006).
Murtaugh, L. C. Pancreas and beta-cell development: from the actual to the possible. Development 134, 427–438 (2007).
Suzuki, K. et al. Transcriptional regulatory factor X6 (Rfx6) increases gastric inhibitory polypeptide (GIP) expression in enteroendocrine K-cells and is involved in GIP hypersecretion in high fat diet-induced obesity. J. Biol. Chem. 288, 1929–1938 (2013).
Kitamura, K. et al. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat. Genet. 32, 359–369 (2002).
Lee, K., Mattiske, T., Kitamura, K., Gecz, J. & Shoubridge C. Reduced polyalanine-expanded Arx mutant protein in developing mouse subpallium alters Lmo1 transcriptional regulation. Hum. Mol. Genet. 23, 1084–1094 (2014).
Jackson, R. S. et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet. 16, 303–306 (1997).
O'Rahilly, S. et al. Brief report: impaired processing of prohormones associated with abnormalities of glucose homeostasis and adrenal function. N. Engl. J. Med. 333, 1386–1390 (1995).
Martin, M. G. et al. Congenital proprotein convertase 1/3 deficiency causes malabsorptive diarrhea and other endocrinopathies in a pediatric cohort. Gastroenterology 145, 138–148 (2013).
Bandsma, R. H. et al. From diarrhea to obesity in prohormone convertase 1/3 deficiency: age-dependent clinical, pathologic, and enteroendocrine characteristics. J. Clin. Gastroenterol. 47, 834–843 (2013).
Yourshaw, M. et al. Exome sequencing finds a novel PCSK1 mutation in a child with generalized malabsorptive diarrhea and diabetes insipidus. J. Pediatr. Gastroenterol. Nutr. 57, 759–767 (2013).
Barzaghi, F., Passerini, L. & Bacchetta, R. Immune dysregulation, polyendocrinopathy, enteropathy, x-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front. Immunol. 3, 211 (2012).
Lampasona, V. et al. Autoantibodies to harmonin and villin are diagnostic markers in children with IPEX syndrome. PLoS ONE 8, e78664 (2013).
Passerini, L. et al. CD4+ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci. Transl. Med. 5, 215ra174 (2013).
McMurchy, A. N. et al. A novel function for FOXP3 in humans: intrinsic regulation of conventional T cells. Blood 121, 1265–1275 (2013).
Horino, S. et al. Selective expansion of donor-derived regulatory T cells after allogeneic bone marrow transplantation in a patient with IPEX syndrome. Pediatr. Transplant. 18, E25–E30 (2014).
Scaillon, M. et al. Severe gastritis in an insulin-dependent child with an IPEX syndrome. J. Pediatr. Gastroenterol. Nutr. 49, 368–370 (2009).
Hashimura Y. et al. Minimal change nephrotic syndrome associated with immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Pediatr. Nephrol. 24, 1181–1186 (2009).
Verbsky, J. W. & Chatila, T. A. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases. Curr. Opin. Pediatr. 25, 708–714 (2013).
Liu, L. et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J. Exp. Med. 208, 1635–1648 (2011).
Alangari, A. et al. LPS-responsive beige-like anchor (LRBA) gene mutation in a family with inflammatory bowel disease and combined immunodeficiency. J. Allergy Clin. Immunol. 130, 481–488 (2012).
Charbonnier, L. M. et al. Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like disorder caused by loss-of-function mutations in LRBA. J. Allergy Clin. Immunol. 135, 217–227 (2015).
Glocker, E. O. et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N. Engl. J. Med. 361, 2033–2045 (2009).
Zigmond, E. et al. Macrophage-restricted interleukin-10 receptor deficiency, but not IL-10 deficiency, causes severe spontaneous colitis. Immunity 40, 720–733 (2014).
Shouval, D. S. et al. Interleukin-10 receptor signalling in innate immune cells regulates mucosal immune tolerance and anti-inflammatory macrophage function. Immunity 40, 706–719 (2014).
Engelhardt, K. R. et al. Clinical outcome in IL-10-and IL-10 receptor-deficient patients with or without hematopoietic stem cell transplantation. J. Allergy Clin. Immunol. 131, 825–830 (2013).
Bacchetta, R. et al. Immunological outcome in haploidentical-HSC transplanted patients treated with IL-10-anergized donor T cells. Front. Immunol. 5, 16 (2014).
Castaldo, G., Lembo, F. & Tomaiuolo, R. Molecular diagnostics: between chips and customized medicine. Clin. Chem. Lab. Med. 48, 973–982 (2010).
Maruotti, G. M. et al. Prenatal diagnosis of inherited diseases: 20 years' experience of an Italian Regional Reference Centre. Clin. Chem. Lab. Med. 51, 2211–2217 (2013).
Berni Canani, R. et al. Genotype-dependency of butyrate efficacy in children with congenital chloride diarrhea. Orphanet. J. Rare Dis. 8, 194 (2013).
1000 Genomes. 1000 Genomes [online], (2014).
Lancaster, M. A. & Knoblich, J. A. Organogenesis in a dish: modeling development and disease using organoid technologies. Science 345, 1247125 (2014).
Passerini, L., Sio, F. R., Porteus, M. H. & Bacchetta, R. Gene/cell therapy approaches for immune dysregulation polyendocrinopathy enteropathy X-linked syndrome. Curr. Gene Ther. 14, 422–428 (2014).
Agne, M. et al. Modularized CRISPR/dCas9 effector toolkit for target-specific gene regulation. ACS Synth. Biol. 3, 986–989 (2014).
Giordano, S. et al. Molecular and functional analysis of the large 5′ promoter region of CFTR gene revealed pathogenic mutations in CF and CFTR-related disorders. J. Mol. Diagn. 15, 331–340 (2013).
Amato, F. et al. Gene mutation in microRNA target sites of CFTR gene: a novel pathogenetic mechanism in cystic fibrosis? PLoS ONE 8, e60448 (2013).
Amato, F. et al. Design, synthesis and biochemical investigation, by in vitro luciferase report system, of peptide nucleic acids as a new inhibitors of mirR-509-3p involved in the regulation of cystic fibrosis disease-gene expression. Med. Chem. Comm. 5, 68–71 (2014).
Ashworth, I., Wilson, A., Hii, M., Macdonald, S. & Hill, S. Long-term outcome of intestinal epithelial cell dysplasia/tufting enteropathy. J. Pediatr. Gastroenterol. Nutr. 58, 239 (2014).
International Microvillus Inclusion Disease (MVID) Patient Registry. International Microvillus Inclusion Disease (MVID) Patient Registry [online].
Congenital Diarrheal Disorders. CongenitalDiarrhealDisorders.net [online].
IPEX Syndrome Consortium. IPEX Syndrome Consortium [online].
Baum, M. et al. Nucleotide sequence of the Na+/H+ exchanger-8 in patients with congenital sodium diarrhea. J. Pediatr. Gastroenterol. Nutr. 53, 474–477 (2011).
Walters, J. R. F. Bile acid diarrhoea and FGF19: new views on diagnosis, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 11, 426–434 (2014).
Acknowledgements
Grants from Regione Campania, DGRC 1901/09 and Agenzia Italiana del Farmaco (AIFA) MRAR08W002 (to R.B.C.), and from NIDDK (#DK083762), and CIRM (RT2-01985) (to M.G.M.) and Italian Telethon Foundation (TGT11A4) (to R.B.) are gratefully acknowledged. The authors thank V. Pezzella for help on text editing.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects in the production of this article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Canani, R., Castaldo, G., Bacchetta, R. et al. Congenital diarrhoeal disorders: advances in this evolving web of inherited enteropathies. Nat Rev Gastroenterol Hepatol 12, 293–302 (2015). https://doi.org/10.1038/nrgastro.2015.44
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrgastro.2015.44
This article is cited by
-
Chronische Diarrhö
Pädiatrie (2023)
-
Approach to Congenital Diarrhea and Enteropathies (CODEs)
Indian Journal of Pediatrics (2023)
-
Chronische Diarrhö bei Kindern und Jugendlichen
Gastro-News (2022)
-
Intestinal immunoregulation: lessons from human mendelian diseases
Mucosal Immunology (2021)
-
Congenital chloride diarrhea clinical features and management: a systematic review
Pediatric Research (2021)
