
Key Points
-
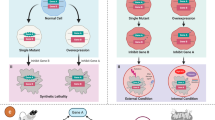
Synthetic lethal genetic interactions with tumour-specific mutations may be exploited to develop anticancer therapeutics.
-
Synthetic dosage lethality and conditional synthetic lethality can expand the scope of conventional synthetic lethal studies.
-
Genetic interaction networks in model organisms provide a framework for screening cancer-relevant candidate synthetic lethal interactions in human cells.
-
Large-scale screening for cancer gene-specific synthetic lethal candidates in human cells has progressed through advances in RNA interference and the CRISPR–Cas9 system.
-
The CRISPR–Cas9 technology is a versatile platform for exploring genetic networks and synthetic lethal interaction phenotypes.
-
The search for synthetic lethality-based therapeutic strategies could be enhanced by integrating synthetic lethal interactions from three distinct sources: model organism genetic networks, human high-throughput screening and synthetic lethal predictions from statistical genetics.
Abstract
A synthetic lethal interaction occurs between two genes when the perturbation of either gene alone is viable but the perturbation of both genes simultaneously results in the loss of viability. Key to exploiting synthetic lethality in cancer treatment are the identification and the mechanistic characterization of robust synthetic lethal genetic interactions. Advances in next-generation sequencing technologies are enabling the identification of hundreds of tumour-specific mutations and alterations in gene expression that could be targeted by a synthetic lethality approach. The translation of synthetic lethality to therapy will be assisted by the synthesis of genetic interaction data from model organisms, tumour genomes and human cell lines.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
References
Hyman, D. M., Taylor, B. S. & Baselga, J. Implementing genome-driven oncology. Cell 168, 584–599 (2017).
Laskin, J. et al. Lessons learned from the application of whole-genome analysis to the treatment of patients with advanced cancers. Cold Spring Harb. Mol. Case Stud. 1, a000570 (2015).
Stockley, T. L. et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med. 8, 109 (2016).
Swanton, C. et al. Consensus on precision medicine for metastatic cancers: a report from the MAP conference. Ann. Oncol. 27, 1443–1448 (2016).
Pagliarini, R., Shao, W. & Sellers, W. R. Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep. 16, 280–296 (2015).
Dobzhansky, T. Genetics of natural populations; recombination and variability in populations of Drosophila pseudoobscura. Genetics 31, 269–290 (1946).
Lucchesi, J. C. Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanogaster. Genetics 59, 37–44 (1968).
Kaiser, C. A. & Schekman, R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell 61, 723–733 (1990).
Hennessy, K. M., Lee, A., Chen, E. & Botstein, D. A group of interacting yeast DNA replication genes. Genes Dev. 5, 958–969 (1991).
Bender, A. & Pringle, J. R. Use of a screen for synthetic lethal and multicopy suppressee mutants to identify two new genes involved in morphogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 11, 1295–1305 (1991).
Nagel, R., Semenova, E. A. & Berns, A. Drugging the addict: non-oncogene addiction as a target for cancer therapy. EMBO Rep. 17, 1516–1531 (2016).
Hartwell, L. H., Szankasi, P., Roberts, C. J., Murray, A. W. & Friend, S. H. Integrating genetic approaches into the discovery of anticancer drugs. Science 278, 1064–1068 (1997). This study suggests that model organism genetics can be used to identify drug targets in human cancer and is the first to propose synthetic lethality screening as a strategy to develop anticancer therapeutics.
Zack, T. I. et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 (2013).
Yan, H., Gibson, S. & Tye, B. K. Mcm2 and Mcm3, two proteins important for ARS activity, are related in structure and function. Genes Dev. 5, 944–957 (1991).
Kroll, E. S., Hyland, K. M., Hieter, P. & Li, J. J. Establishing genetic interactions by a synthetic dosage lethality phenotype. Genetics 143, 95–102 (1996).
Bian, Y. et al. Synthetic genetic array screen identifies PP2A as a therapeutic target in Mad2-overexpressing tumors. Proc. Natl Acad. Sci. USA 111, 1628–1633 (2014).
Reid, R. J. et al. A synthetic dosage lethal genetic interaction between CKS1B and PLK1 is conserved in yeast and human cancer cells. Genetics 204, 807–819 (2016).
Duffy, S. et al. Overexpression screens identify conserved dosage chromosome instability genes in yeast and human cancer. Proc. Natl Acad. Sci. USA 113, 9967–9976 (2016).
Tong, A. H. et al. Global mapping of the yeast genetic interaction network. Science 303, 808–813 (2004).
Haber, J. E. et al. Systematic triple-mutant analysis uncovers functional connectivity between pathways involved in chromosome regulation. Cell Rep. 3, 2168–2178 (2013).
Bunting, S. F. et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254 (2010).
Jaspers, J. E. et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 3, 68–81 (2013).
Bindra, R. S. et al. Down-regulation of Rad51 and decreased homologous recombination in hypoxic cancer cells. Mol. Cell. Biol. 24, 8504–8518 (2004).
Chan, N. et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 70, 8045–8054 (2010).
Bandyopadhyay, S. et al. Rewiring of genetic networks in response to DNA damage. Science 330, 1385–1389 (2010). This study describes the creation of condition- dependent genetic interaction maps in yeast and demonstrates that genetic interactions can change in response to DNA damage.
Guenole, A. et al. Dissection of DNA damage responses using multiconditional genetic interaction maps. Mol. Cell 49, 346–358 (2013).
Li, X., O'Neil, N. J., Moshgabadi, N. & Hieter, P. Synthetic cytotoxicity: digenic interactions with TEL1/ATM mutations reveal sensitivity to low doses of camptothecin. Genetics 197, 611–623 (2014).
Rouleau, M., Patel, A., Hendzel, M. J., Kaufmann, S. H. & Poirier, G. G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 10, 293–301 (2010).
Bailey, M. L. et al. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Mol. Cancer Ther. 13, 724–732 (2014).
Hartwell, L. H. & Kastan, M. B. Cell cycle control and cancer. Science 266, 1821–1828 (1994).
Rudrapatna, V. A., Cagan, R. L. & Das, T. K. Drosophila cancer models. Dev. Dyn. 241, 107–118 (2012).
Kirienko, N. V., Mani, K. & Fay, D. S. Cancer models in Caenorhabditis elegans. Dev. Dyn. 239, 1413–1448 (2010).
Tarailo, M., Tarailo, S. & Rose, A. M. Synthetic lethal interactions identify phenotypic “interologs” of the spindle assembly checkpoint components. Genetics 177, 2525–2530 (2007).
Dixon, S. J., Andrews, B. J. & Boone, C. Exploring the conservation of synthetic lethal genetic interaction networks. Commun. Integr. Biol. 2, 78–81 (2009).
Boucher, B. & Jenna, S. Genetic interaction networks: better understand to better predict. Front. Genet. 4, 290 (2013).
Baryshnikova, A., Costanzo, M., Myers, C. L., Andrews, B. & Boone, C. Genetic interaction networks: toward an understanding of heritability. Annu. Rev. Genomics Hum. Genet. 14, 111–133 (2013).
Roguev, A. et al. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science 322, 405–410 (2008).
Ryan, C. J. et al. Hierarchical modularity and the evolution of genetic interactomes across species. Mol. Cell 46, 691–704 (2012).
McLellan, J. L. et al. Synthetic lethality of cohesins with PARPs and replication fork mediators. PLoS Genet. 8, e1002574 (2012).
Tong, A. H. et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 (2001).
Pan, X. et al. A robust toolkit for functional profiling of the yeast genome. Mol. Cell 16, 487–496 (2004).
Costanzo, M. et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 353, aaf1420 (2016). This study describes a global genetic interaction network derived from the analysis of pairwise interactions of nearly all the 6,000 genes in S. cerevisiae.
McManus, K. J., Barrett, I. J., Nouhi, Y. & Hieter, P. Specific synthetic lethal killing of RAD54B-deficient human colorectal cancer cells by FEN1 silencing. Proc. Natl Acad. Sci. USA 106, 3276–3281 (2009).
Srivas, R. et al. A network of conserved synthetic lethal interactions for exploration of precision cancer therapy. Mol. Cell 63, 514–525 (2016). This study describes a large-scale cross-species approach to identify clinically relevant genetic interactions with genes mutated in cancer.
Fire, A. et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391, 806–811 (1998).
Elbashir, S. M. et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 (2001).
Paddison, P. J., Caudy, A. A., Bernstein, E., Hannon, G. J. & Conklin, D. S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 16, 948–958 (2002).
Berns, K. et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 428, 431–437 (2004).
Paddison, P. J. et al. A resource for large-scale RNA-interference-based screens in mammals. Nature 428, 427–431 (2004).
Cheung, H. W. et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl Acad. Sci. USA 108, 12372–12377 (2011).
Vizeacoumar, F. J. et al. A negative genetic interaction map in isogenic cancer cell lines reveals cancer cell vulnerabilities. Mol. Syst. Biol. 9, 696 (2013).
Bhinder, B. & Djaballah, H. Systematic analysis of RNAi reports identifies dismal commonality at gene-level and reveals an unprecedented enrichment in pooled shRNA screens. Comb. Chem. High Throughput Screen. 16, 665–681 (2013).
Reynolds, A. et al. Rational siRNA design for RNA interference. Nat. Biotechnol. 22, 326–330 (2004).
Fellmann, C. et al. Functional identification of optimized RNAi triggers using a massively parallel sensor assay. Mol. Cell 41, 733–746 (2011).
Jackson, A. L. et al. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 21, 635–637 (2003).
Birmingham, A. et al. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat. Methods 3, 199–204 (2006).
Sigoillot, F. D. et al. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat. Methods 9, 363–366 (2012).
Buehler, E. et al. siRNA off-target effects in genome-wide screens identify signaling pathway members. Sci. Rep. 2, 428 (2012).
Hoffman, G. R. et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl Acad. Sci. USA 111, 3128–3133 (2014).
Bassik, M. C. et al. Rapid creation and quantitative monitoring of high coverage shRNA libraries. Nat. Methods 6, 443–445 (2009).
Downward, J. RAS synthetic lethal screens revisited: still seeking the elusive prize? Clin. Cancer Res. 21, 1802–1809 (2015).
Haibe-Kains, B. et al. Inconsistency in large pharmacogenomic studies. Nature 504, 389–393 (2013).
Haverty, P. M. et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 533, 333–337 (2016).
Iorio, F. et al. A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754 (2016).
Cong, L. et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013).
Jinek, M. et al. RNA-programmed genome editing in human cells. eLife 2, e00471 (2013).
Mali, P. et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826 (2013).
Shalem, O., Sanjana, N. E. & Zhang, F. High-throughput functional genomics using CRISPR-Cas9. Nat. Rev. Genet. 16, 299–311 (2015).
Aguirre, A. J. et al. Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 6, 914–929 (2016).
Munoz, D. M. et al. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discov. 6, 900–913 (2016).
Wang, T. et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101 (2015).
Hart, T. et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526 (2015).
Horlbeck, M. A. et al. Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5, e19760 (2016). This study introduces the second generation of genome-wide CRISPRi and CRISPRa libraries.
Wang, T. et al. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell 168, 890–903 (2017). This study is one of several recent papers that use a new generation of genome-wide CRISPR knockout libraries to search for cell line-dependent genetic vulnerabilities and potential synthetic lethality candidates.
Blomen, V. A. et al. Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096 (2015).
Horlbeck, M. A. et al. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. eLife 5, e12677 (2016).
Kuscu, C., Arslan, S., Singh, R., Thorpe, J. & Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 32, 677–683 (2014).
Qi, L. S. et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013).
Gilbert, L. A. et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013).
Gilbert, L. A. et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661 (2014).
Konermann, S. et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588 (2015).
Wong, A. S. et al. Multiplexed barcoded CRISPR-Cas9 screening enabled by CombiGEM. Proc. Natl Acad. Sci. USA 113, 2544–2549 (2016). This study combines the method CombiGEM with the CRISPR–Cas9 system to enable multiplexed synthetic lethality screening strategies.
Kabadi, A. M., Ousterout, D. G., Hilton, I. B. & Gersbach, C. A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 42, e147 (2014).
Sakuma, T., Nishikawa, A., Kume, S., Chayama, K. & Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 4, 5400 (2014).
Han, K. et al. Synergistic drug combinations for cancer identified in a CRISPR screen for pairwise genetic interactions. Nat. Biotechnol. 35, 463–474 (2017).
Shen, J. P. et al. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat. Methods 14, 573–576 (2017).
Wong, A. S., Choi, G. C., Cheng, A. A., Purcell, O. & Lu, T. K. Massively parallel high-order combinatorial genetics in human cells. Nat. Biotechnol. 33, 952–961 (2015).
Dixit, A. et al. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866 (2016).
Adamson, B. et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882 (2016). References 88 and 89 introduce the method of Perturb-seq, which combines CRISPR-based screening with single-cell transcriptional readout for a highly integrated phenotypic analysis of genetic perturbations.
Jaitin, D. A. et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167, 1883–1896 (2016).
Datlinger, P. et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301 (2017).
Leiserson, M. D., Wu, H. T., Vandin, F. & Raphael, B. J. CoMEt: a statistical approach to identify combinations of mutually exclusive alterations in cancer. Genome Biol. 16, 160 (2015).
Babur, O. et al. Systematic identification of cancer driving signaling pathways based on mutual exclusivity of genomic alterations. Genome Biol. 16, 45 (2015).
Zhang, F. et al. Predicting essential genes and synthetic lethality via influence propagation in signaling pathways of cancer cell fates. J. Bioinform. Comput. Biol. 13, 1541002 (2015).
Wappett, M. et al. Multi-omic measurement of mutually exclusive loss-of-function enriches for candidate synthetic lethal gene pairs. BMC Genomics 17, 65 (2016).
Jackson, R. A. & Chen, E. S. Synthetic lethal approaches for assessing combinatorial efficacy of chemotherapeutic drugs. Pharmacol. Ther. 162, 69–85 (2016).
Jerby-Arnon, L. et al. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell 158, 1199–1209 (2014). The authors developed and applied the statistical model DAISY, which analyses and integrates cancer genomic data and experimental genetic screens to predict synthetic lethal candidates.
Guo, J., Liu, H. & Zheng, J. SynLethDB: synthetic lethality database toward discovery of selective and sensitive anticancer drug targets. Nucleic Acids Res. 44, D1011–D1017 (2016).
Thoma, C. R. et al. A high-throughput-compatible 3D microtissue co-culture system for phenotypic RNAi screening applications. J. Biomol. Screen. 18, 1330–1337 (2013).
Matano, M. et al. Modeling colorectal cancer using CRISPR-Cas9-mediated engineering of human intestinal organoids. Nat. Med. 21, 256–262 (2015).
Bruna, A. et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell 167, 260–274 (2016).
Braun, C. J. et al. Versatile in vivo regulation of tumor phenotypes by dCas9-mediated transcriptional perturbation. Proc. Natl Acad. Sci. USA 113, E3892–E3900 (2016).
Chen, S. et al. Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260 (2015).
Walcott, F. L., Patel, J., Lubet, R., Rodriguez, L. & Calzone, K. A. Hereditary cancer syndromes as model systems for chemopreventive agent development. Semin. Oncol. 43, 134–145 (2016).
Wu, X. & Lippman, S. M. An intermittent approach for cancer chemoprevention. Nat. Rev. Cancer 11, 879–885 (2011).
Bryant, H. E. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917 (2005).
Farmer, H. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921 (2005). References 106 and 107 show the first evidence of a synthetic lethality-based therapeutic strategy between the BRCA genes, which are commonly mutated in breast and other cancers, and PARP1.
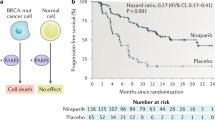
Fong, P. C. et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 361, 123–134 (2009).
Gelmon, K. A. et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 12, 852–861 (2011).
Mateo, J. et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 373, 1697–1708 (2015).
McCabe, N. et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 66, 8109–8115 (2006).
Lord, C. J. & Ashworth, A. BRCAness revisited. Nat. Rev. Cancer 16, 110–120 (2016).
Barber, L. J. et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 229, 422–429 (2013).
Ter Brugge, P. et al. Mechanisms of therapy resistance in patient-derived xenograft models of BRCA1-deficient breast cancer. J. Natl Cancer Inst. 108, djw148 (2016).
Xu, G. et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521, 541–544 (2015).
Rottenberg, S. et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc. Natl Acad. Sci. USA 105, 17079–17084 (2008).
Knezevic, C. E. et al. Proteome-wide profiling of clinical PARP inhibitors reveals compound-specific secondary targets. Cell Chem. Biol. 23, 1490–1503 (2016).
Schiewer, M. J. & Knudsen, K. E. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res. 12, 1069–1080 (2014).
Burkle, A. & Virag, L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol. Aspects Med. 34, 1046–1065 (2013).
Murai, J. et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 13, 433–443 (2014).
Hopkins, T. A. et al. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol. Cancer Res. 13, 1465–1477 (2015).
Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol. Oncol. 5, 387–393 (2011).
Acknowledgements
P.H. is a senior fellow in the Genetic Networks program at the Canadian Institute for Advanced Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Synthetic lethality
-
A synthetic lethal interaction occurs between two genes when a perturbation (a mutation, RNA interference knockdown or inhibition) that affects either gene alone is viable but the perturbation of both genes simultaneously is lethal.
- Non-homologous end-joining
-
(NHEJ). The repair of double-strand DNA breaks by direct ligation without the use of a homologous template.
- Homologous recombination
-
The exchange of nucleotide sequences between identical (or near identical) DNA molecules. Homologous recombination is the most common form of homology-directed repair of double-strand DNA breaks.
- Genetic interaction network
-
A genetic interaction occurs when the perturbation of one or more genes affects the phenotype of another gene alteration. A genetic interaction network defines the functional relationship between many genes.
- Orthologue
-
Genes in different species that are originated from a single gene of the last common ancestor.
- Modules
-
Groups of genes or proteins that act together in a common cellular function.
- Synthetic sickness
-
A synthetic sickness interaction occurs between two genes when a perturbation (a mutation, RNA interference knockdown or inhibition) that affects either gene alone is viable but the disruption of both genes simultaneously results in a reduction of viability.
- Driver mutations
-
Mutations that confer a selective growth advantage to a cancer or a pre-cancerous cell.
- Isogenic
-
Organisms or cell lines that contain identical or nearly identical genotypes.
- Heterogeneity
-
In a cell line or tumour, the diversity of genotypes within the population.
- Gene essentiality profiles
-
Sets of genes required for proliferation or viability in the context of a single cell line or tumour type.
Rights and permissions
About this article

Cite this article
O'Neil, N., Bailey, M. & Hieter, P. Synthetic lethality and cancer. Nat Rev Genet 18, 613–623 (2017). https://doi.org/10.1038/nrg.2017.47
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrg.2017.47
This article is cited by
-
Leveraging synthetic lethality to uncover potential therapeutic target in gastric cancer
Cancer Gene Therapy (2024)
-
A replication competent Plasmodium falciparum parasite completely attenuated by dual gene deletion
EMBO Molecular Medicine (2024)
-
Uncovering hidden cancer self-dependencies through analysis of shRNA-level dependency scores
Scientific Reports (2024)
-
Genome-wide CRISPR screens identify novel regulators of wild-type and mutant p53 stability
Molecular Systems Biology (2024)
-
Clinical application of PARP inhibitors in ovarian cancer: from molecular mechanisms to the current status
Journal of Ovarian Research (2023)
