
Abstract
In countries with the best cancer outcomes, approximately 60% of patients receive radiotherapy as part of their treatment, which is one of the most cost-effective cancer treatments. Notably, around 40% of cancer cures include the use of radiotherapy, either as a single modality or combined with other treatments. Radiotherapy can provide enormous benefit to patients with cancer. In the past decade, significant technical advances, such as image-guided radiotherapy, intensity-modulated radiotherapy, stereotactic radiotherapy, and proton therapy enable higher doses of radiotherapy to be delivered to the tumour with significantly lower doses to normal surrounding tissues. However, apart from the combination of traditional cytotoxic chemotherapy with radiotherapy, little progress has been made in identifying and defining optimal targeted therapy and radiotherapy combinations to improve the efficacy of cancer treatment. The National Cancer Research Institute Clinical and Translational Radiotherapy Research Working Group (CTRad) formed a Joint Working Group with representatives from academia, industry, patient groups and regulatory bodies to address this lack of progress and to publish recommendations for future clinical research. Herein, we highlight the Working Group's consensus recommendations to increase the number of novel drugs being successfully registered in combination with radiotherapy to improve clinical outcomes for patients with cancer.
Similar content being viewed by others

Main
Although cancer mortality is declining in some European Union countries, lifestyle factors and the ageing population mean that cancer incidence will continue to rise1. Cancer survival gains will result from improved curative treatment2, and among those cancer patients who are cured, it has been estimated that 49% are cured by surgery, 40% by radiotherapy alone or combined with other modalities, and 11% by chemotherapy alone or combined with other modalities3. Radiotherapy is also a highly effective treatment for palliation and symptom control in patients with advanced-stage or recurrent cancer4.
Radiotherapy is a very cost-effective component of cancer care. The estimated total cost of radiotherapy in the UK was only 5% of the estimated total cost of cancer care, which is representative of other developed countries5,6. Significant technical advances, such as image-guided radiotherapy, intensity-modulated radiotherapy, stereotactic radiotherapy, and proton therapy have enabled, when compared with conventional radiotherapy, the application of higher radiation doses and more precise cancer targeting with considerably lower doses to non-malignant surrounding tissues. These advances have improved patient outcomes7,8. Moreover, there is clear (Level 1) evidence that drug-radiotherapy combinations improve overall survival9. Apart from the combination of traditional cytotoxic chemotherapy with radiotherapy, however, limited progress has been made in using potential synergies between targeted systemic therapies and radiotherapy. Specifically, very few new drug–radiotherapy combinations are registered10,11.
There is an unmet need for intelligent and rational approaches to drug–radiotherapy combinations on the basis of our molecular understanding of radiobiology and increased ability to develop agents that can be combined with radiotherapy in preclinical models. To define the most rational clinical approaches, the National Cancer Research Institute Clinical and Translational Radiotherapy Research Working Group (CTRad) formed a Working Group with representatives from academia, industry, patient groups and regulatory bodies to address this lack of progress and to publish recommendations for future research and development. This article explains the consensus recommendations of the CTRad Working Group, which aim to increase the number of novel agents being successfully registered in combination with radiotherapy to improve outcomes for patients with cancer.
Methods
CTRad was established in 2009 by the UK National Cancer Research Institute (NCRI) to focus on clinical and translational issues relating to radiotherapy and radiobiology. The CTRad Working Group has developed a portfolio of practice-changing trials and actively promotes translation of new discoveries into practice12. In September 2014, the CTRad Working Group formed at a meeting in London, UK, to identify both barriers and solutions to increase the number of clinical trials of drug–radiotherapy combinations and to design realistic registration strategies for these combinations. The organizing committee approached all pharmaceutical and biotechnology companies in a list of contacts collated by the Experimental Cancer Medicine Centres (ECMC) and Cancer Research UK (CRUK) Combinations Alliance. The Working Group decided to provide clear guidelines for researchers and pharmaceutical companies working in the field, by publishing a Consensus Statement to highlight important opinions about the issue of drug–radiotherapy combinations, and recommend a particular course of action. Based on the summary of the discussions at that meeting, the issues were divided into eight topics, with the intention of agreeing eight consensus recommendations. It was made clear to the Working Group members that the consensus recommendations would be eminence-based, and should present a balanced view of the field based on the current evidence base.
The follow-up meeting in London in September 2015 was held with the specific intention of reaching consensus on the eight recommendations from the previous year. Invitations were sent to all Working Group members who had attended the previous meeting, plus additional members of the radiotherapy community who had shown interest in discussions at CTRad meetings subsequent to the previous meeting, as well as additional biotechnology and pharmaceutical companies contacted via the ECMC/CRUK Combinations Alliance, and regulatory contacts at the European Medicines Agency (EMA) and Medicines and Health products Regulatory Agency (MHRA). The aim was to be as broad and inclusive as possible to maximise the impact of the recommendations from the Working Group.
Of the 54 people invited to the meeting in September 2015, 37 attended and several others contributed with comments on the draft consensus statements and figures, before and after the meeting. All delegates worked in small groups to agree on the eight consensus statements: drug–radiotherapy combinations; route to registration; clinical trial end points; changing the standard of care; clinical trial methodology; radiotherapy quality assurance; preclinical dataset and target population; and patient and consumer involvement to raise awareness (Box 1). All delegates rotated through all eight groups. All authors were given the opportunity to comment on the manuscript and approve the final version before publication. Agreement was reached for the Joint Working Group to reconvene in 5–10 years from the publication of this article in order to assess progress achieved in the advancement of drug–radiotherapy combinations.
Consensus recommendations
Drug–radiotherapy combinations
Principles of radiobiology. Ionizing radiation is most often delivered as photons in the X-ray wavelength of the electro-magnetic spectrum, but it can also be delivered as particles in the form of electrons or protons. The effect of radiotherapy includes direct damage to DNA in cells, or indirect damage caused by the X-rays colliding with molecules within the target, from primary and secondary ionisation events. For example, free electrons cause damage to molecules other than DNA within the cell or in other cells, or can interact with water molecules, leading to the generation of hydroxyl radicals or other reactive oxygen species13. Conventional radiotherapy is given with curative intent in fractions of 1.8–2.0 Gy daily on weekdays up to 35 times. The purpose of fractionation is to maximize the killing of cancer cells, while minimizing effects on normal tissue in and around the target volume. This concept is called the Therapeutic Index (Fig. 1). Hypofractionation refers to giving a lower number of fractions larger than 2.0 Gy, which is more effective per unit dose owing to the curvature of the ionizing radiation dose–response curve, but carries greater risk of toxicity to non-malignant tissues. Several large-scale clinical trials have found that giving a higher (>2.0 Gy) dose of radiotherapy per fraction, and a fewer (<35) number of fractions in total, can be as safe as conventional radiotherapy, resulting in a change in the standard of care for common cancers, such as breast cancer and prostate cancer14,15.

A variety of strategies have shown promise in ameliorating ionizing radiation damage to normal tissues, including protection with radical scavengers, stimulating recovery with cytokines, modifying the p53 response, reducing the negative effects of inflammatory cascades and oxidative stress, and the use of stem-cell therapy. Of note, the slopes of clinical dose–response curves (the relationship between the probability of tumour control and the ionizing radiation dose) indicate that increasing the effective ionizing radiation dose by just 10% (a dose enhancement factor of 1.1) will increase tumour control rates by 5–30%, depending on the tumour site and whether control rates are already low or high121,122.
The lethal effects of radiotherapy primarily arise from damage to DNA. Radiation-induced DNA lesions include base pair damage, single-strand breaks (SSBs) and double-strand breaks (DSBs), which are considered to be the most lethal. SSBs are more rapidly repaired by cells than the DSBs, which are more likely to cause mutagenesis or lethality. Approximately, 1 Gy of photon radiotherapy results in 1 × 105 ionization events per cell, producing 1,000–2,000 SSBs and 40 DSBs, with the majority of DSB repair occurring within the first 2 h of the fraction of radiotherapy16.
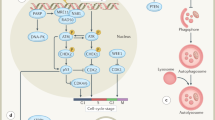
Certain biological features of tumours can affect outcomes after radiotherapy; for example, the extent and degree of hypoxia17, the ability of the surviving cells to repopulate within the treatment time (typically 6–7 weeks for conventionally fractionated radiotherapy)18, and the intrinsic radioresistance of the tumour cells19. In addition, the microenvironment, the immune environment, and cellular energetics can also affect responses to radiotherapy (Fig. 2), which illustrates the multiple biological consequences of radiotherapy.

Irradiation of the tumour causes a variety of biological consequences, which can be exploited by combining radiotherapy with novel agents that target the relevant pathways123. ATR, ataxia telangiectasia and Rad3-related protein; CA9, carbonic anhydrase 9; Chk1, checkpoint kinase 1; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; DDR, DNA damage response; DNA-PK, DNA-dependent protein kinase; HIF-1-α, hypoxia-inducible factor 1-alpha; MCT 1, monocarboxylate transporter 1; MCT 4, monocarboxylate transporter 4; mTOR, mechanistic target of rapamycin ; PARP, poly(ADP-ribose) polymerase; PD-1, programmed cell death protein 1; PI3K, phosphoinositide 3-kinase; NF-κB, nuclear factor-kappa-B; UPR, unfolded protein response.
Novel drug–radiotherapy combinations. Combining novel drugs with radiotherapy has clear potential to considerably improve patient outcomes. Moreover, several agents in development target each of the radiobiological effect categories (Fig. 2). Collaboration between industry and academia is essential for progress in this field, and should occur as early as possible when a new drug is being developed. For industry to invest in new drug–radiotherapy combinations, a robust scientific basis for the combination in preclinical models needs to be demonstrated, and a route of registration must be defined for each drug–radiotherapy combination in terms of patient selection and clinical trial end points.
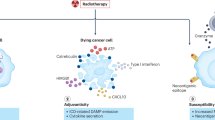
Proposed drug–radiotherapy combinations should have a sound scientific basis with regards to radiobiology, immuno-oncology, molecular biology and pharmacology. Traditionally, strategies for combining drugs with radiotherapy have focused either on hypoxia modification (Box 2) or on altering the intrinsic radiosensitivity of the irradiated tumour(s) within the target volume for radiotherapy (Box 3). Local control of the tumour is not the only end point to consider when designing a drug–radiotherapy combination strategy (Fig. 2). Radiation-induced bystander effects are biological effects caused in cells that have not been directly irradiated20. Such effects include DNA damage, chromosomal instability, mutation, and the induction of apoptosis21. For example, irradiation of the tumour microenvironment (that is, within and around the tumour) might be an important determinant of the efficacy of radiotherapy22.
A growing interest is placed in combining radiotherapy with immunotherapy23,24.This particular drug–radiotherapy combination is emerging as a new field of research, termed immuno-radio-oncology (Box 4). Ionising radiation causes immunogenic cell death of cancer cells, modulates antigen presentation by cancer cells and alters the microenvironment within the irradiated field25,26,27. Importantly, this approach might promote enhanced anticancer responses to a systemic drug therapy, such as a monoclonal antibody against cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)28. Local radiotherapy can enhance responses to immune-modulating agents at sites distant from irradiated areas. This phenomenon is known as the abscopal effect, and represents an important and exciting development in the potential role and scope of radiation therapy, as demonstrated in an important proof-of-principle clinical trial with results published in 2015 (Ref. 29). In the next few years, improvements in viral and bacterial gene-delivery systems30,31 and in oncolytic virotherapy vectors32 might result in significant advances in the safety and efficacy of gene and viral therapies to target the interaction between cancer cells and their microenvironment33.
The route to registration
Existing regulatory guidance on the development of chemotherapy and radiotherapy combinations is very limited, and early discussion with and scientific advice from regulatory agencies is recommended through pre-Investigational New Drug (IND) discussions. The Committee for Medicinal Products for Human Use (CHMP) Guideline on the evaluation of anticancer medicinal products in man acknowledges the importance of combination therapy by combining compounds with non-overlapping toxicities and/or mechanisms of resistance or activity34. The CHMP guideline mentions the use of radiotherapy and/or chemotherapy sensitisers; however, given the limited regulatory experience of such combinations, early interactions with regulators need to be undertaken. The FDA published guidance on co-development of two or more IND for use in combination35. Although the European Medicines Agency CHMP guideline does not address the specific issue of the combination of drugs and radiotherapy, it is highly relevant in the development of combination therapies in general and provides a helpful framework for discussions with the FDA, EMA and other regulators.
Oncology drug development differs from most other therapy areas in the early involvement of patients (rather than healthy volunteers), usually from first-in-human dose-finding study development onwards, and in the wide range of academic and collaborative groups involved in partnership with the drug developer. This scenario should provide early opportunities for consideration of combinations of radiotherapy with chemotherapy in patients with cancer. We suggest that, in cases with a good biological and therapeutic rationale, studies on radiotherapy combinations should be considered as part of the design of early-phase studies in patients (Fig. 3). These protocols might be designed in an adaptive manner to support the early initiation of combinations once the maximum tolerated dose (MTD) or biologically effective dose (BED) for a single agent has been established. A clinical development plan for a new drug–radiotherapy combination should define 'go/no go' and potential acceleration criteria (biomarker/efficacy/toxicity) for each decision point, which can be the move from preclinical to phase I, the end of the dose escalation part of a phase I study, any cohort expansions in phase I, or the move to phase II and phase III trials36.

Early interactions with regulatory agencies are recommended because of the limited published regulatory guidance. CHMP, Committee for Medicinal Products for Human Use; EMA, European Medicines Agency; IND, investigational new drug; MOA, mechanism of action; MTD, maximum tolerated dose; NDA, new drug application; PI/PII/PIII, phase of development of clinical trial; RT, radiotherapy.
The early introduction of radiotherapy combinations, either simultaneously or soon after initial marketing authorisation, is dependent on the early initiation of combination modality studies and a clear pathway to registration. Ideally, the initiation of combination studies should involve agreement on the tumour site, clinical end points to be studied and acceptable clinical trial designs from phase I to III. The apparent lack of a clear pathway to registration might be a disincentive for the developer to undertake early studies. Regulatory agencies, however, provide numerous opportunities to engage and encourage development of drug–modality combinations34,37. For example, EMA Scientific Advice can be sought at any stage of development and can provide the study sponsor with access to a European network of both academic and regulatory experts through Scientific Advice Groups (SAG), which include SAG-Oncology. Such discussions might include the review of the biological rationale, clinical study designs or relevant clinical study end points. Similar opportunities are provided by the FDA and other national regulators. Such advice is applicable to drug–radiotherapy development as well as to new drug–drug combinations.
Given the high number of unmet needs in oncology, new agents might possibly be approved under accelerated regulatory procedures, such as the FDA accelerated approval scheme, which allows a new drug approval owing to intermediate end points — provided subsequent confirmatory studies are ongoing to study conventional, long-term end points. The EMA Priority Medicines scheme proposed in 2015 will provide “enhanced scientific and regulatory support to companies developing new therapeutic options to patients who currently have no treatment options, or a major therapeutic advantage over existing treatments” (Ref. 38). Early engagement with regulators, the academic community, patients and stakeholders should occur in the development of new therapies to ensure that access to new treatments and consequent health improvements are achieved in the shortest possible time frame.
The commercial opportunities of novel treatment combinations cannot be overlooked (Box 5). These opportunities include repurposing drugs to be used as radiosensitisers, for which extensive phase I to IV clinical experience of the drug might already exist from their use in other indications39. The development route for a new drug could include changes to the patent life of candidate drugs when they are combined with radiotherapy, that is, a new indication for the drug. Data exclusivity, market protection or patent extensions might be sought on the basis of orphan designation or new therapeutic claims in many jurisdictions, including the USA and European Union (EU)40,41.
Clinical end points
Outside of the palliative setting, the aim of radiotherapy treatment is to achieve local and regional tumour control, either as the primary treatment modality, or as adjuvant or neoadjuvant therapy in combination with surgery. In the absence of metastatic disease, locoregional control should translate into improvements in disease-free survival and/or overall survival. In the presence of micrometastatic disease, locoregional tumour control might still translate into improvements in overall survival, symptom-free survival and quality of life (Table 1).
The selection of the most meaningful clinical end point(s) requires consideration of several factors, which include the tumour site, disease stage and target patient population. Early or intermediate end points can add predictive value, particularly in the context of cancers with a better prognosis. The pathological complete response (pCR) is the best validated end point for some solid cancers in the neoadjuvant setting, such as rectal cancer and breast cancer42,43; nevertheless, this surrogate end point is not measurable in all disease sites and standardisation of histopathology procedures requires careful quality assurance.
When considering drug-radiotherapy combinations, end points that enable evaluation of non-malignant tissue toxicity should be included to ensure that improvements in tumour control and survival end points do not occur at the expense of unacceptable increases in such toxicities. Guidance on dose constraints for organs at risk (OARs) is based on historical data for radiotherapy alone; international guidance should be followed in assessing non-malignant tissue toxicities for new drug–radiotherapy combinations (Box 6). Whereas the studies from Emami et al.44 have historically been used to estimate the dose of radiotherapy deliverable to non-malignant tissues when radiotherapy is used alone, these conservative estimates are not specifically applicable to drug–radiotherapy combinations except as a general guide. The need for a systematic approach to data collection on non-malignant tissue toxicities is emphasized by the QUANTEC reviews45. For example, in one article from this series, the authors emphasize that the biological determinants of the risk of non-malignant tissue toxicity vary between individuals, and the factors that influence it are specific to a given radiation pathogenesis46. This is also the case for drug–radiotherapy combinations, emphasising the need for the collection of accurate data in order to inform predictive models that might be developed in the future.
The addition of concomitant cytotoxic chemotherapy to radical radiotherapy has resulted in increases in overall survival and/or disease-free survival in a broad range of tumour types (such as cancers of the head and neck, lung, cervix, rectum, glioblastoma) and addition of molecular targeted agents to radiotherapy has improved overall survival and disease-free survival in head and neck cancer47,48. Regarding the interests of individuals with cancer, regulatory agencies increasingly recognize the value of patient-reported outcomes as end points of clinical trials49.
Changing the standard of care
For a drug–radiotherapy combination treatment to change clinical practice, it is paramount that the combination has demonstrated improved outcomes over the existing standard of care. This standard of care can vary geographically, and might depend on each country's judgement of factors, such as clinical efficacy, cost-effectiveness and the balance of toxicities of alternative treatment options. Ideally, investigators should ensure that the results of the proposed clinical development plan are applicable widely, rather than specific to a particular geographical region; for example, acknowledging the widespread use in most countries of platinum-based chemotherapy concomitantly with radiotherapy for non-small-cell lung cancer (NSCLC) or head and neck cancer50. Indeed, one challenge for investigators is that clinical trial designs might need to consider how to incorporate data on the changing standard of care during the study progression.
The key objective of phase I studies of new potential anticancer treatments is to determine a recommended phase II dose (RP2D) to take forward into further clinical studies. The fundamental assumption that has traditionally underpinned drug development in oncology is that 'more is better'; that is, the higher the dose, the greater the potential for antitumour activity. This dogma has resulted in the RP2D of anticancer treatments usually being the MTD — the highest dose of a drug or treatment that does not cause unacceptable adverse effects51. This concept is not necessarily applicable to molecularly targeted agents, which are associated with different adverse effects to cytotoxic chemotherapy drugs, as lower drug doses than the MTD can be used to achieve synergy for some molecularly targeted agents52. In situations in which synergy is expected from the combination of two treatment modalities, the minimal biologically effective dose (MBED) of one or both of the treatments might be lower than that required for that agent administered as monotherapy53,54. Clinically relevant antitumour activity (the MBED, or even the maximum therapeutic effect (MTE)), might occur at doses significantly lower than the MTD55. The ability to use drugs at lower doses in drug–radiotherapy combinations than when used as a single agent, and possibly for shorter periods of time, can reduce drug costs for the combined treatment modality, with potentially fewer adverse effects than with the drug alone.
One option to allow delivery of a drug–radiotherapy combination in a timely manner is to conduct the dose-escalation part of phase I studies of the drug as a single agent alongside studies of the drug–radiotherapy combination. Data on safety, tolerability and pharmacokinetic (PK) end points from each dose level of the single agent study can be incorporated into the parallel study of the drug–radiotherapy combination. The choice of dose(s) to be explored further with radiotherapy are likely to be driven by modelling of the clinical PK data with the preclinical efficacy studies to define the expected minimal biologically active dose (MBAD) and, if relevant and feasible, tumour tissue sampling of putative biomarkers of the MBED (such as serological markers, circulating tumour cells or imaging readouts), which can be mandated in all patients during the dose escalation part of the clinical trial56. These approaches are likely to find the correlation between dose and proof-of-mechanism (PoM) or proof-of-principle (PoP) biomarkers, giving a better assessment of MBAD57. However, it should be noted that the inherent variability of these biomarkers might require more than 3–6 patients per cohort for a meaningful result to be obtained.
Phase I oncology studies frequently include small expansion cohorts to explore biological activity; the end points can be clinical, such as objective response rate (ORR), or biomarker-driven, such as paired tumour biopsies for PoM or PoP biomarkers. The doses explored in these expansion cohorts are usually at, or close to, the MTD, but rarely allow determination of MBED. A more robust approach is to consider expanding cohorts to determine MBAD and test at least one dose between MBAD and MTD; such an approach might be triggered by significant tumour responses seen in the MBAD phase.
Clinical trial methodology
Early-phase trials are required to determine the optimal way to give a novel drug in combination with radiotherapy. Several types of trial design exist and are used according to the specific setting and research question under evaluation47,58.
Acute toxicity from radiotherapy is not generally a good predictor of late toxic effects, with the exception of particularly severe or durable acute reactions that can have consequential late effects. On account of the risk of sub-acute or late toxicities from radiotherapy, clinical trials of drug–radiotherapy combinations might require extended follow-up periods, or monitoring of patients beyond the routine follow-up period (Box 7). Extending the cohort follow-up period before dose escalation would provide a more robust RP2D, and could minimize delay by the use of a modification to the continual reassessment method (CRM), the time-to-event (TITE)-CRM59. This method incorporates late toxicity events that emerge after the typical short-term dose-limiting toxicity (DLT) period. The TITE-CRM includes the TITE for each patient — the event being a DLT and therefore including data from patients whose required follow-up period has not yet completed — and also allows the potential for patients to be recruited continuously in parallel60. Other adaptive study designs might enable more robust RP2D decisions than those determined using more traditional trial designs, thereby reducing overall timelines to reach the primary end point61.
Clinical trial designs that incorporate measures of both efficacy and toxicity to identify the correct dose62, might offer advantages over traditional dose-escalation designs that identify only the MTD. Alternatively, in settings where the risk of toxicity to the OAR with a drug–radiotherapy combination can vary, alternative approaches to dose cohort allocation should be considered, such as stratification by risk group or isotoxic radiotherapy dosing. Isotoxic dosing allows for personalised radiotherapy by maximising dose to tumour while remaining within pre-defined normal tissue constraints. This potentially identifies subgroups of patients more likely to respond to therapy and consequently informs the design of subsequent phase II and/or phase III trials. Early-phase trials of isotoxic dosing in NSCLC have demonstrated promising survival results with limited toxicity63,64. Further trials looking at isotoxic intensity- modulated radiation therapy (IMRT) in NSCLC are focusing on feasibility, MTD and toxicity65. Research into newer techniques such as stereotactic ablative radiotherapy might benefit from using isotoxic dose prescription derived from experiences of conventionally fractionated radiotherapy66.
As more drug–radiotherapy combinations are developed, the need to consider more than simply drug-dose escalation will arise. Emerging methodologies developed for drug–drug combinations could be applied to drug–radiotherapy combinations for which the 'dose' of radiotherapy might relate to scheduling or sequencing of the drug–radiotherapy combination (Box 8). Multidimensional approaches, such as the proposed Product of Independent beta Probabilities dose Escalation (PIPE) design, could be used to identify the most promising combinations to take forward to phase II trials67. PIPE is an example of a non-parametric design for a dual agent clinical trial, in which the model parameters are the probabilities of toxicity for each of the dose combinations. Some prior knowledge of likely toxicities is required to create the model, which can then be rapidly updated when toxicity data are obtained in the clinical trial. Alternatively, in cases in which multiple drugs might be combined with radiotherapy for the first time, and recruitment is likely to be steady, cohort allocation could take the form of a FLIP-FLOP design. In this design, dose escalation of each drug occurs in alternate cohorts of patients, enabling continued recruitment during the toxicity observation period of the previous cohort and maximising patient recruitment rates47.
Scenarios in which a run-in period of single agent can precede a drug–radiotherapy combination would support efficient evaluation of any changes in the pharmacokinetic and/or pharmacodynamic (PK/PD) effects of the drug in the presence of radiotherapy. Enrichment trial designs could be employed to further develop biomarkers or to support appropriate phase III trial design68,69,70. Tissue, biofluid or imaging biomarkers have an important role in drug development, particularly in preclinical studies and in early-phase clinical trials71. Biomarkers can enable the identification of potentially successful drugs early on, thus accelerating market approval for some therapies. Biomarkers can also enable the identification of ineffective or toxic compounds at the earliest opportunity72. Relatively few combination studies, particularly combinations of drug–radiotherapy, have incorporated biomarkers to alter decision making in early-phase studies, and this represents an important area for future research. Earlier consideration of potential radiotherapy-related biomarkers or biomarkers associated with mechanisms underpinning a synergistic drug–radiotherapy combination might help reduce the risk of failure in the phase III setting. 'Window of opportunity' clinical trials permit the development of tissue and imaging biomarkers in a short period of time before surgery or other definitive treatment73,74. As gains in the therapeutic index can be achieved by improving efficacy or reducing toxicity, phase II designs that support co-primary end points could be useful75.
A key component of successful drug–radiotherapy combinations is the use of appropriate, robust end points at each phase of assessment. To increase the likelihood of successful phase III trials, decision-making in phase II trials should focus on such end points that provide reliable information for 'go/no-go' decisions. Data on late-stage effects should be pooled, which will require collaboration between both academic and pharmaceutical partners to share trial data76.
Radiotherapy quality assurance
Adequate quality assurance of treatment delivery within clinical trials is critical to the success of drug–radiotherapy studies77. The methodology is well-established for drugs (manufacture, storage, distribution, dosing and compliance) and it should be matched by the accuracy of dose delivery from the radiotherapy equipment. Quality assurance of other steps in the process of radiotherapy treatment, which is partly individualised for each patient based on their anatomy, has been harder to achieve than quality assurance of treatment delivery.
As a starting point, the numerous radiotherapy-quality assurance (RTQA) procedures and naming conventions used globally have been harmonized in a new naming convention to be used in clinical studies incorporating radiation therapy78. This overview will facilitate intergroup study collaboration, simplifying exchange and interpretation of RTQA results (Box 9). The recommendation is for this process to start early in the development of drug–radiotherapy combinations, to ensure the radiotherapy component can be adopted widely if later phase trials are pursued. Streamlining of the RTQA process for these later phase trials, with a centralised credentialing process, is pursued on a trial-by-trial basis. Streamlining prevents clinical sites and central groups from being overburdened, and ensures that the level of RTQA is consistent with the level of risk and the clinical end points of the study.
The electronic data from every patient treated within every trial is collated by the UK's National Cancer Research Institute's Radiotherapy Trials Quality Assurance group (NCRI RTTQA) group to allow retrospective auditing of protocol adherence. This provides an analysable dataset to explore relationships between delivered radiation dose and acute or late organ-specific toxicity and tumour control. Data from previous trials can be used to guide future trials if patients can be stratified, allowing different cohorts to be escalated in parallel.
Preclinical dataset and target population
Appropriate preclinical translational studies are essential to provide data that enables the most promising novel drug–radiotherapy combinations to be identified and progressed. The data should support regulatory approval and inform the optimal clinical development programme. The preclinical dataset should address four important concepts: first, demonstrate that the novel drug improves the efficacy of radiotherapy in clinically relevant models (both in vitro and in vivo); second, define an effective dose schedule; third, provide an assessment of non-malignant tissue toxicity for the drug–radiotherapy combination to identify potential clinical risks; and fourth, identify potential responsive patient subpopulations and possible candidate biomarkers.
Before designing an early-phase clinical study, investigators must define the proposed treatment in instances of unmet need, and select a registerable end point that the clinical study will pursue. This will inform on the tissue type and end point for the models used in the preclinical phase. For example, if the standard of clinical care is cisplatin-based chemoradiotherapy, preclinical modelling should ensure that antagonism does not occur when the new drug is added to cisplatin-based chemoradiation. While in vitro studies should initially be used to examine drug–radiotherapy combinations, in vivo tumour models are likely to be more informative. There has been interest in using mouse models that might be more molecularly diverse or possess more complex stroma, such as patient-derived xenografts (PDX) or genetically engineered mouse models (GEMs)79,80. These models have considerable time/resource considerations; whether data derived from studies with these models would provide substantially more information is unclear. However, although cell line-derived tumour xenografts remain the principal model for examining efficacy, there can be significant value in establishing the effect of an intact host immune response on the efficacy of the drug–radiotherapy combination and indeed, for combinations of immunotherapies (Box 4) and radiotherapy, syngeneic and immunocompetent mouse models are essential. Ideally, the use of small irradiators in studies involving animals would match the approach to be applied clinically81,82.
As an end point for efficacy studies, tumour cure has been considered as the gold standard for assessing the impact of drug–radiotherapy combinations. Tumour cure is the most comprehensive method, but the generation of TCD50 values (the dose of radiation required to cure 50% of animals) with and without drug is extremely labour intensive and impractical for most laboratories. In addition, there are examples in which TCD50 data can give a distorted view of the value of drug–radiotherapy combinations. In some studies, the addition of EGFR pathway inhibitors to radiation treatment had no effect on TCD50, yet substantial growth delays were observed preclinically, and clinical benefit was observed with the combination83,84,85.
A growth-delay end point, which evaluates the time taken for tumours to reach a defined volume (commonly a trebling or quadrupling in volume from treatment initiation), is more achievable than tumour cure and should be used routinely. After irradiation, tumours will generally regrow at the same rate as unirradiated tumours. Therefore, by analysing the rate of regrowth after treatment, important mechanistic information related to drug activity can be obtained — including the magnitude and duration of tumour regression in relation to pretreatment tumour volume. These experiments should quantify the improvement in the tumour response to radiation derived from adding the drug. For example, to determine if less radiotherapy can be delivered in the presence of the drug to be isoeffective, tumour–dose responses for radiotherapy alone must be established. We also recommend examining a novel drug–radiotherapy combination in a minimum of two relevant tumour models, and ideally examining a fractionated radiotherapy schedule.
Efficacy studies should assess drug sequencing in relation to the timing of radiotherapy treatment. If the drug is given before radiotherapy, it might function to condition the microenvironment (such as reduce hypoxia), to synchronize cells or to inhibit an immediate response target. The drug might need, however, to be present at different stages to increase the magnitude of radiation damage (for example, a hypoxic cell radiosensitiser). In some cases, the drug might need to be present after irradiation to inhibit repair or to drive cells down an apoptotic, autophagic or mitotic cell-death pathway. Scheduling experiments will be enhanced by a clear understanding of the PK/PD properties of the drug, so that rational dosing can be applied during fractionated radiotherapy. Design of subsequent clinical work will then need to take account of PK/PD observations from first-in-human studies.
A mechanistic rationale will usually indicate why tumours should be more affected than non-malignant tissue by the combined treatment in the irradiation field. Currently, there is no formal prerequisite for combination toxicology studies to support registration. However, given that drug treatment also has the potential to augment both the early and long-term toxic effects of radiotherapy, the examination of these effects in preclinical models is considered a prudent measure for new mechanistic classes of drug and those of the same class with differing PK and selectivity profiles.
A preliminary assessment of the skin can be made in the proximity of the irradiated tumour86,87,88,89,90,91. Furthermore, acute skin responses can be predictive of late toxicity90,91. Other non-malignant tissue assays can be prioritised based on the tumour site of interest, such as lung pneumonitis and fibrosis models for drug–radiotherapy combinations used to treat lung cancers, or mucositis models in the case of gastrointestinal tumours92,93,94,95,96. These studies must include radiation-only dose responses so that any enhancement of toxicity can be interpreted meaningfully, ideally with the same drug dose or radiation dose schedules used in complementary efficacy experiments. These studies should then give an indication of whether the efficacy of radiotherapy has been preferentially enhanced compared with toxicity, to produce a discernable increase in the therapeutic index. Current challenges for conducting such studies include a lack of standardisation of protocols and limited reporting of data. Future studies should aim to benchmark the effects of such combinations in model systems to provide guidance on the relevance of findings generated with new drug–radiotherapy combinations.
Novel oncology drugs that enter clinical evaluation are now developed from the outset with a strong hypothesis for the subset of cancer patients most likely to respond to treatment. Studies will incorporate the use of exploratory PD and patient selection biomarkers, the latter having the potential to be developed further as a companion diagnostic.
A pragmatic approach to patient selection can be to examine whether the relevant tumour types also represent an established setting for the use of radiotherapy (Table 1). However, a mechanistic basis might exist for an enhanced therapeutic effect in the combination of a particular signalling inhibitor with radiotherapy, even if the inhibitor has little disease modifying activity alone. Such an effect can be obtained if the drug treatment modulates a key factor that limits the effectiveness of radiotherapy (such as DNA repair, cell-cycle phase redistribution, tumour reoxygenation, cellular repopulation, and intrinsic radiosensitivity; Fig. 1)97. Furthermore, the magnitude of enhancement can be dependent on tumour type, and might provide a patient stratification hypothesis for the combination. Given the potential for mechanistic interactions between novel drugs and radiotherapy, preclinical studies should be used to carefully assess combination regimens, using tumour models that are linked to a clinical development strategy. Clinically derived 'signatures' of prognosis following radical radiotherapy, on the basis of genomic and microenvironmental indices, might also be considered for use in patient selection once these biomarkers have been validated in prospective clinical trials98,99.
Finally, it is important to verify that the PD biomarkers being used for drug evaluation are not adversely influenced by radiotherapy treatment. Additional biomarkers relevant to radiotherapy (DNA-damage end points, tumour vascular perfusion, or tumour hypoxia, among others) should be examined, with further elaboration using intravital imaging100. Inclusion of these end points can provide further mechanistic insight into the effects of combination treatment, and might influence the choice of methods incorporated into early clinical studies.
Patient and consumer involvement to raise awareness
Within the UK, patients, carers and others affected by cancer (consumers) are invited to participate in all aspects of the NCRI's work. All consumers are members of the NCRI Consumer Forum, allowing exchange of knowledge and expertise in a coordinated way. In 2015, the NCRI Consumer Forum stated its guiding principle of “working together to build a community with the common purpose of providing patient and public perspectives throughout the research process, to deliver research with better outcomes and experiences for all” (Ref. 101). The involvement of patients in trial design is associated with recognized benefits; patients have better outcomes in research-active centres than in other centres, and patients involved in research know more about their condition than patients without that involvement102. The experience and expertise of patients and carers is unique, and can inform on trial design. This opportunity enables patients to be part of the solution to the problems faced by researchers when designing a trial. Radiotherapy and drug–radiotherapy combination clinical trials, however, bring with them added complexity. Patients might experience increased risk derived from these therapies than from standard therapy and, of course, each patient and their family will be bringing their own attitude to risk, their context and their values into the trial.
The fear of the unknown is present in situations in which the patient might already be feeling vulnerable and with decreased hope. The experienced utility (what the patient is living with) and imagined disutility (what might happen) affect patients; mis-imagining the future state has a significant bearing on decision-making. Clinicians bring what is possible to the discussion; the patient brings their preference and what is valuable to them as individuals. To overcome potential barriers, patients need transparency. They need to know about local control end points and end points recognized by regulatory authorities. They need to know about de-escalation, organ-sparing, tumour shrinkage and control, patient-related outcomes and quality of life. They need to be involved as early as possible in the design of the trial so that there are patient advocates who can defend and justify the trial. They require a discourse that first presents the benefits, followed by the risks. They also need support and coaching to enable them to live with uncertainty and accept their situation.
Discussion
Improving long-term control rates and overall survival rates from cancer is of significant societal benefit. We have illustrated that relatively short treatment periods for drug–radiotherapy combinations, or relatively modest changes to the therapeutic index, have the potential to be cost-effective and meaningful for society. Combinations of molecularly targeted drugs with radiotherapy have generally failed to improve overall survival rates for the small number of cancers studied with a dual modality approach. Therefore, a robust scientific rationale to support new drug–radiotherapy combinations is of great importance. The preclinical package of data required to justify new combinations and to reduce risk from an industry perspective has been outlined in this document. Collaboration between academia and industry, and funding in public–private partnership (including academic entrepreneurship) are essential to the success of drug–radiotherapy combinations, both in preclinical development and in the clinical development plan for a new drug. The new research field of immuno-radio-oncology is likely to dramatically broaden the scope of radiotherapy in the future, beyond its use to achieve local control, organ sparing or cure, by the use of radiotherapy to stimulate responses to systemic therapy. One notable example is the abscopal effect.
We have deliberately challenged the view sometimes expressed in industry that the route to registration for a drug–radiotherapy combination is likely to be long and arduous10. Using positive examples, such as the global harmonisation of RTQA and the ability to include radiotherapy in existing clinical trial designs, we have demonstrated the importance of considering this particular combination treatment as early as possible during the development plan for a new drug. In addition to the well-established role of radiotherapy in treating over half of patients diagnosed with cancer, drug–radiotherapy combinations offer significant potential for improving therapy outcomes. Involving patients early in the process, and having honest conversations and explaining the benefits followed by the risks are a first step to achieving this goal. Based on our collective experience, we encourage investigators to follow the guidelines set out in this Consensus Statement in order to increase the number of novel drugs being successfully registered in combination with radiotherapy to improve clinical outcomes for patients with cancer.
Conclusions
There is an unmet need for rational approaches to drug–radiotherapy combinations based on molecular understanding of radiobiology and our increasing ability to translate the most promising results from preclinical model systems. The National Cancer Research Institute Clinical and Translational Radiotherapy Research Working Group (CTRad) formed a Joint Working Group with representatives from academia, industry, patient groups and regulatory bodies to address the recent lack of progress in the field of drug–radiotherapy combinations and to publish recommendations for future research and development. The Working Group decided to divide the courses of action required by investigators in this field in to eight topics, and they consequently agreed eight eminence-based consensus recommendations (Box 1). The aim of this article, and the consensus recommendations contained within it, is to increase the number of novel agents being successfully registered in combination with radiotherapy to improve outcomes for patients with cancer. The Joint Working Group will reconvene in 5–10 years from publication of this article in order to assess progress achieved in the advancement of drug–radiotherapy combinations.
References
Stewart, B. W. & Wild, C. P. World Cancer Report (World Health Organisation Press, 2014).
Department of Health. Radiotherapy in England. GOV.UK https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/213151/Radiotherapy-Services-in-England-2012.pdf (2012).
Ringborg, U. et al. The Swedish Council on Technology Assessment in Health Care (SBU) systematic overview of radiotherapy for cancer including a prospective survey of radiotherapy practice in Sweden 2001 — summary and conclusions. Acta Oncol. 42, 357–365 (2003).
Lutz, S. T., Jones, J. & Chow, E. Role of radiation therapy in palliative care of the patient with cancer. J. Clin. Oncol. 32, 2913–2919 (2014).
Cullen, J., Drabble, D., Castellanos, C. & Brissett, L. Recommendations for achieving a world-class radiotherapy service in the UK. The Tavistock Institute http://www.tavinstitute.org/wp-content/uploads/2014/05/Tavistock_Projects_Recommendations-for-achieving-a-world-class-radiotherapy-service-in-the-UK-.pdf (2014).
Atun, R. et al. Expanding global access to radiotherapy. Lancet Oncol. 16, 1153–1186 (2015).
Tree, A. C. et al. Stereotactic body radiotherapy for oligometastases. Lancet Oncol. 14, e28–e37 (2013).
Chang, J. Y., Senan, S., Smit, E. F. & Roth, J. A. Surgery versus SABR for resectable non-small-cell lung cancer — authors' reply. Lancet Oncol. 16, e374–e375 (2015).
Lawrence, Y. R. et al. NCIRTOG translational program strategic guidelines for the early-stage development of radiosensitizers. J. Natl Cancer Inst. 105, 11–24 (2013).
Ataman, O. U. et al. The clinical development of molecularly targeted agents in combination with radiation therapy: a pharmaceutical perspective. Int. J. Radiat. Oncol. Biol. Phys. 84, e447–e454 (2012).
Glass, C., Den, R. B., Dicker, A. P. & Lawrence, Y. R. Toxicity of phase I radiation oncology trials: worldwide experience. Int. J. Radiat. Oncol. Biol. Phys. 78, S65–S65 (2010).
National Cancer Research Institute Clinical Studies Groups. Portfolio maps. NCRI Clinical Studies Groups http://csg.ncri.org.uk/portfolio/portfolio-maps/ (2016).
Niederhuber, J. E., Armitage, J. O., Doroshow, J. H., Kastan, M. B. & Tepper, J. E. Abeloff's Clinical Oncology (Elsevier Health Sciences, 2013).
Haviland, J. S. et al. The UK Standardisation of Breast Radiotherapy (START) trials of radiotherapy hypofractionation for treatment of early breast cancer: 10-year follow-up results of two randomised controlled trials. Lancet Oncol. 14, 1086–1094 (2013).
Dearnaley, D. et al. Conventional versus hypofractionated high-dose intensity-modulated radiotherapy for prostate cancer: preliminary safety results from the CHHiP randomised controlled trial. Lancet Oncol. 13, 43–54 (2012).
Lewanski, C. R. & Gullick, W. J. Radiotherapy and cellular signalling. Lancet Oncol. 2, 366–370 (2001).
Nordsmark, M. et al. Prognostic value of tumor oxygenation in 397 head and neck tumors after primary radiation therapy. An international multi-center study. Radiother. Oncol. 77, 18–24 (2005).
Kim, J. J. & Tannock, I. F. Repopulation of cancer cells during therapy: an important cause of treatment failure. Nat. Rev. Cancer 5, 516–525 (2005).
West, C. M., Davidson, S. E., Roberts, S. A. & Hunter, R. D. Intrinsic radiosensitivity and prediction of patient response to radiotherapy for carcinoma of the cervix. Br. J. Cancer 68, 819–823 (1993).
Butterworth, K. T., McMahon, S. J., Hounsell, A. R., O'Sullivan, J. M. & Prise, K. M. Bystander signalling: exploring clinical relevance through new approaches and new models. Clin. Oncol. 25, 586–592 (2013).
Marin, A. et al. Bystander effects and radiotherapy. Rep. Pract. Oncol. Radiother. 20, 12–21 (2015).
Barcellos-Hoff, M. H., Park, C. & Wright, E. G. Radiation and the microenvironment — tumorigenesis and therapy. Nat. Rev. Cancer 5, 867–875 (2005).
Demaria, S., Golden, E. B. & Formenti, S. C. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 1, 1325–1332 (2015).
Illidge, T. Turning radiotherapy into an effective systemic anti-cancer treatment in combination with immunotherapy. Clin. Oncol. (R. Coll. Radiol.) 27, 696–699 (2015).
Thompson, R. F. & Maity, A. Radiotherapy and the tumor microenvironment: mutual influence and clinical implications. Adv. Exp. Med. Biol. 772, 147–165 (2014).
Yoshimura, M., Itasaka, S., Harada, H. & Hiraoka, M. Microenvironment and radiation therapy. Biomed. Res. Int. 2013, 685308 (2013).
Shiao, S. L. & Coussens, L. M. The tumor-immune microenvironment and response to radiation therapy. J. Mammary Gland Biol. Neoplasia 15, 411–421 (2010).
Dewan, M. Z. et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin. Cancer Res. 15, 5379–5388 (2009).
Golden, E. B. et al. Local radiotherapy and granulocyte-macrophage colony-stimulating factor to generate abscopal responses in patients with metastatic solid tumours: a proof-of-principle trial. Lancet Oncol. 16, 795–803 (2015).
Hingorani, M. et al. Combining radiation and cancer gene therapy: a potential marriage of physical and biological targeting? Curr. Cancer Drug Targets 7, 389–409 (2007).
Mengesha, A. et al. Potential and limitations of bacterial-mediated cancer therapy. Front. Biosci. 12, 3880–3891 (2007).
Harrington, K. J. et al. Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin. Cancer Res. 16, 4005–4015 (2010).
Barker, H. E., Paget, J. T., Khan, A. A. & Harrington, K. J. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat. Rev. Cancer 15, 409–425 (2015).
European Medicines Agency. Guideline on the evaluation of anticancer medicinal products in man. European Medicines Agency http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2013/01/WC500137128.pdf (2012).
U.S. Food and Drug Administration. Guidance for industry: codevelopment of two or more new investigational drugs for use in combination. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm236669.pdf (2013).
Cook, D. et al. Lessons learned from the fate of AstraZeneca's drug pipeline: a five-dimensional framework. Nat. Rev. Drug Discov. 13, 419–431 (2014).
Medicines and Healthcare Products Regulatory Agency. Marketing authorisations, variations and licensing guidance. GOV.UK https://www.gov.uk/topic/medicines-medical-devices-blood/marketing-authorisations-variations-licensing (2016).
European Medicines Agency. Priority medicines (PRIME) scheme. http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_000660.jsp&mid= (2015).
Hill, E. J. et al. Clinical trial of oral nelfinavir before and during radiation therapy for advanced rectal cancer. Clin. Cancer Res. 22, 1922–1931 (2016).
U.S. Food and Drug Administration. Frequently asked questions on patents and exclusivity. http://www.fda.gov/Drugs/DevelopmentApprovalProcess/ucm079031.htm (2014).
European Medicines Agency. Data exclusivity, market protection and paediatric rewards. http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2013/05/WC500143122.pdf (2013)
Maas, M. et al. Long-term outcome in patients with a pathological complete response after chemoradiation for rectal cancer: a pooled analysis of individual patient data. Lancet Oncol. 11, 835–844 (2010).
Le Scodan, R. et al. Breast cancer with synchronous metastases: survival impact of exclusive locoregional radiotherapy. J. Clin. Oncol. 27, 1375–1381 (2009).
Emami, B. et al. Tolerance of normal tissue to therapeutic irradiation. Int. J. Radiat. Oncol. Biol. Phys. 21, 109–122 (1991).
Bentzen, S. M. et al. Quantitative Analyses of Normal Tissue Effects in the Clinic (QUANTEC): an introduction to the scientific issues. Int. J. Radiat. Oncol. Biol. Phys. 76, S3–S9 (2010).
Bentzen, S. M. et al. Biomarkers and surrogate endpoints for normal-tissue effects of radiation therapy: the importance of dose-volume effects. Int. J. Radiat. Oncol. Biol. Phys. 76, S145–S150 (2010).
Harrington, K. J. et al. Guidelines for preclinical and early phase clinical assessment of novel radiosensitisers. Br. J. Cancer 105, 628–639 (2011).
Seiwert, T. Y., Salama, J. K. & Vokes, E. E. The concurrent chemoradiation paradigm — general principles. Nat. Clin. Pract. Oncol. 4, 86–100 (2007).
Di Maio, M., Basch, E., Bryce, J. & Perrone, F. Patient-reported outcomes in the evaluation of toxicity of anticancer treatments. Nat. Rev. Clin. Oncol. 13, 319–325 (2016).
Ang, K. K. et al. Randomized phase III trial of concurrent accelerated radiation plus cisplatin with or without cetuximab for stage III to IV head and neck carcinoma: RTOG 0522. J. Clin. Oncol. 32, 2940–2950 (2014).
National Cancer Institute. NCI Dictionary of Cancer Terms. http://www.cancer.gov/publications/dictionaries/cancer-terms?cdrid=546597 (2016).
Glynne-Jones, R., Dunst, J. & Sebag-Montefiore, D. The integration of oral capecitabine into chemoradiation regimens for locally advanced rectal cancer: how successful have we been? Ann. Oncol. 17, 361–371 (2006).
Chou, T. C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 70, 440–446 (2010).
Tallarida, R. J. Quantitative methods for assessing drug synergism. Genes Cancer 2, 1003–1008 (2011).
Jain, R. K. et al. Phase I oncology studies: evidence that in the era of targeted therapies patients on lower doses do not fare worse. Clin. Cancer Res. 16, 1289–1297 (2010).
O'Connor, J. P., Jackson, A., Parker, G. J., Roberts, C. & Jayson, G. C. Dynamic contrast-enhanced MRI in clinical trials of antivascular therapies. Nat. Rev. Clin. Oncol. 9, 167–177 (2012).
Smethurst, D. & Hughes, A. A. Proposed structure to classify levels of proof within a clinical development programme. Int. J. Pharm. Med. 19, 227–232 (2005).
Pijls-Johannesma, M. et al. A systematic methodology review of phase I radiation dose escalation trials. Radiother. Oncol. 95, 135–141 (2010).
Cheung, Y. K. & Chappell, R. Sequential designs for phase I clinical trials with late-onset toxicities. Biometrics 56, 1177–1182 (2000).
Desai, S. P. et al. Phase I study of oxaliplatin, full-dose gemcitabine, and concurrent radiation therapy in pancreatic cancer. J. Clin. Oncol. 25, 4587–4592 (2007).
Le Tourneau, C., Lee, J. J. & Siu, L. L. Dose escalation methods in phase I cancer clinical trials. J. Natl Cancer Inst. 101, 708–720 (2009).
Thall, P. F. & Cook, J. D. Dose-finding based on efficacy-toxicity trade-offs. Biometrics 60, 684–693 (2004).
van Baardwijk, A. et al. Mature results of a phase II trial on individualised accelerated radiotherapy based on normal tissue constraints in concurrent chemo-radiation for stage III non-small cell lung cancer. Eur. J. Cancer 48, 2339–2346 (2012).
De Ruysscher, D. et al. Individualised isotoxic accelerated radiotherapy and chemotherapy are associated with improved long-term survival of patients with stage III NSCLC: a prospective population-based study. Radiother. Oncol. 102, 228–233 (2012).
Christodoulou, M., Bayman, N., McCloskey, P., Rowbottom, C. & Faivre-Finn, C. New radiotherapy approaches in locally advanced non-small cell lung cancer. Eur. J. Cancer 50, 525–534 (2014).
Zindler, J. D. et al. Increasing the therapeutic ratio of stereotactic ablative radiotherapy by individualized isotoxic dose prescription. J. Natl Cancer Inst. 108, djv305 (2015).
Mander, A. P. & Sweeting, M. J. A product of independent beta probabilities dose escalation design for dual-agent phase I trials. Stat. Med. 34, 1261–1276 (2015).
Redig, A. J. & Janne, P. A. Basket trials and the evolution of clinical trial design in an era of genomic medicine. J. Clin. Oncol. 33, 975–977 (2015).
Freidlin, B., McShane, L. M., Polley, M. Y. & Korn, E. L. Randomized phase II trial designs with biomarkers. J. Clin. Oncol. 30, 3304–3309 (2012).
McShane, L. M., Hunsberger, S. & Adjei, A. A. Effective incorporation of biomarkers into phase II trials. Clin. Cancer Res. 15, 1898–1905 (2009).
Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 69, 89–95 (2001).
Yap, T. A., Sandhu, S. K., Workman, P. & de Bono, J. S. Envisioning the future of early anticancer drug development. Nat. Rev. Cancer 10, 514–523 (2010).
Mehta, S. et al. Assessing early therapeutic response to bevacizumab in primary breast cancer using magnetic resonance imaging and gene expression profiles. J. Natl Cancer Inst. Monogr. 2011, 71–74 (2011).
Peeters, S. G. et al. TH-302 in combination with radiotherapy enhances the therapeutic outcome and is associated with pretreatment [18F]HX4 hypoxia PET imaging. Clin. Cancer Res. 21, 2984–2992 (2015).
Tournoux, C., De Rycke, Y., Medioni, J. & Asselain, B. Methods of joint evaluation of efficacy and toxicity in phase II clinical trials. Contemp. Clin. Trials 28, 514–524 (2007).
Taichman, D. B. et al. Sharing clinical trial data — a proposal from the International Committee of Medical Journal Editors. N. Engl. J. Med. 374, 384–386 (2016).
Peters, L. J. et al. Critical impact of radiotherapy protocol compliance and quality in the treatment of advanced head and neck cancer: results from TROG 02.02. J. Clin. Oncol. 28, 2996–3001 (2010).
Melidis, C. et al. Radiation therapy quality assurance in clinical trials — Global Harmonisation Group. Radiother. Oncol. 111, 327–329 (2014).
Hidalgo, M. et al. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 4, 998–1013 (2014).
Sharpless, N. E. & Depinho, R. A. The mighty mouse: genetically engineered mouse models in cancer drug development. Nat. Rev. Drug Discov. 5, 741–754 (2006).
Verhaegen, F., Granton, P. & Tryggestad, E. Small animal radiotherapy research platforms. Phys. Med. Biol. 56, R55–R83 (2011).
Verhaegen, F., van Hoof, S., Granton, P. V. & Trani, D. A review of treatment planning for precision image-guided photon beam pre-clinical animal radiation studies. Z. Med. Phys. 24, 323–334 (2014).
Baumann, M. et al. Selective inhibition of the epidermal growth factor receptor tyrosine kinase by BIBX1382BS and the improvement of growth delay, but not local control, after fractionated irradiation in human FaDu squamous cell carcinoma in the nude mouse. Int. J. Radiat. Biol. 79, 547–559 (2003).
Bonner, J. A. et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 354, 567–578 (2006).
Krause, M. et al. Different classes of EGFR inhibitors may have different potential to improve local tumour control after fractionated irradiation: a study on C225 in FaDu hSCC. Radiother. Oncol. 74, 109–115 (2005).
Denekamp, J., Ball, M. M. & Fowler, J. F. Recovery and repopulation in mouse skin as a function of time after x-irradiation. Radiat. Res. 37, 361–370 (1969).
Rojas, A. et al. Radiosensitisation in normal tissues with oxygen, carbogen or nicotinamide: therapeutic gain comparisons for fractionated x-ray schedules. Radiother. Oncol. 39, 53–64 (1996).
Stewart, F. A., Denekamp, J. & Randhawa, V. S. Skin sensitization by misonidazole: a demonstration of uniform mild hypoxia. Br. J. Cancer 45, 869–877 (1982).
Douglas, B. G. & Fowler, J. F. The effect of multiple small doses of x rays on skin reactions in the mouse and a basic interpretation. Radiat. Res. 66, 401–426 (1976).
Dorr, W. & Hendry, J. H. Consequential late effects in normal tissues. Radiother. Oncol. 61, 223–231 (2001).
Denekamp, J. Early and late radiation reactions in mouse feet. Br. J. Cancer 36, 322–329 (1977).
Citrin, D. E. et al. Role of type II pneumocyte senescence in radiation-induced lung fibrosis. J. Natl Cancer Inst. 105, 1474–1484 (2013).
Moore, B. B. & Hogaboam, C. M. Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L152–L160 (2008).
Giridhar, P., Mallick, S., Rath, G. K. & Julka, P. K. Radiation induced lung injury: prediction, assessment and management. Asian Pac. J. Cancer Prev. 16, 2613–2617 (2015).
Ding, N. H., Li, J. J. & Sun, L. Q. Molecular mechanisms and treatment of radiation-induced lung fibrosis. Curr. Drug Targets 14, 1347–1356 (2013).
Tung, D. et al. Differential effects of cyclosporin and etanercept treatment on various pathologic parameters in a murine model of irradiation-induced mucositis. Curr. Ther. Res. Clin. Exp. 73, 150–164 (2012).
Qayum, N. et al. Tumor vascular changes mediated by inhibition of oncogenic signaling. Cancer Res. 69, 6347–6354 (2009).
Vergis, R. et al. Intrinsic markers of tumour hypoxia and angiogenesis in localised prostate cancer and outcome of radical treatment: a retrospective analysis of two randomised radiotherapy trials and one surgical cohort study. Lancet Oncol. 9, 342–351 (2008).
Lalonde, E. et al. Tumour genomic and microenvironmental heterogeneity for integrated prediction of 5-year biochemical recurrence of prostate cancer: a retrospective cohort study. Lancet Oncol. 15, 1521–1532 (2014).
Palmer, G. M., Fontanella, A. N., Shan, S. & Dewhirst, M. W. High-Resolution In Vivo Imaging of Fluorescent Proteins Using Window Chamber Models (Humana Press, 2012).
National Cancer Research Institute Consumer Forum. Consumer report Pro-forma. http://www.ncri.org.uk/wp-content/uploads/2016/02/CSG-Consumer-Report-September-2015.pdf (2015).
Ozdemir, B. A. et al. Research activity and the association with mortality. PLoS ONE 10, e0118253 (2015).
Jain, R. K. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat. Med. 7, 987–989 (2001).
Nieder, C., Pawinski, A., Dalhaug, A. & Andratschke, N. A review of clinical trials of cetuximab combined with radiotherapy for non-small cell lung cancer. Radiat. Oncol. 7, 3 (2012).
Maier, P., Wenz, F. & Herskind, C. Radioprotection of normal tissue cells. Strahlenther. Onkol. 190, 745–752 (2014).
Postow, M. A. et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N. Engl. J. Med. 366, 925–931 (2012).
Vatner, R. E., Cooper, B. T., Vanpouille-Box, C., Demaria, S. & Formenti, S. C. Combinations of immunotherapy and radiation in cancer therapy. Front. Oncol. 4, 325 (2014).
Stupp, R. et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 10, 459–466 (2009).
Fojo, T. & Grady, C. How much is life worth: cetuximab, non-small cell lung cancer, and the $440 billion question. J. Natl Cancer Inst. 101, 1044–1048 (2009).
Brown, B. et al. An economic evaluation of cetuximab combined with radiotherapy for patients with locally advanced head and neck cancer in Belgium, France, Italy, Switzerland, and the United Kingdom. Value Health 11, 791–799 (2008).
Tsang, Y., Haviland, J., Venables, K. & Yarnold, J. The impact of dose heterogeneity on late normal tissue complication risk after hypofractionated whole breast radiotherapy. Radiother. Oncol. 104, 143–147 (2012).
Ippolito, E. et al. Early proctoscopy is a surrogate endpoint of late rectal toxicity in prostate cancer treated with radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 83, e191–e195 (2012).
Boothe, D. L. et al. Transforming growth factor β-1 (TGF-β1) is a serum biomarker of radiation induced fibrosis in patients treated with intracavitary accelerated partial breast irradiation: preliminary results of a prospective study. Int. J. Radiat. Oncol. Biol. Phys. 87, 1030–1036 (2013).
He, J. et al. The association between TGF-β1 polymorphisms and radiation pneumonia in lung cancer patients treated with definitive radiotherapy: a meta-analysis. PLoS ONE 9, e91100 (2014).
Postel-Vinay, S. et al. Phase I trials of molecularly targeted agents: should we pay more attention to late toxicities? J. Clin. Oncol. 29, 1728–1735 (2011).
Hanna, N. et al. Holland-Frei Cancer Medicine 6th edn (2003).
Fornander, T. et al. Adjuvant tamoxifen in early breast cancer: occurrence of new primary cancers. Lancet 1, 117–120 (1989).
Bresalier, R. S. et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N. Engl. J. Med. 352, 1092–1102 (2005).
Ewer, M. S. & Lippman, S. M. Type II chemotherapy-related cardiac dysfunction: time to recognize a new entity. J. Clin. Oncol. 23, 2900–2902 (2005).
US National Library of Science. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02264678 (2016).
Bentzen, S. M. Radiobiological considerations in the design of clinical trials. Radiother. Oncol. 32, 1–11 (1994).
Diez, P., Vogelius, I. S. & Bentzen, S. M. A new method for synthesizing radiation dose-response data from multiple trials applied to prostate cancer. Int. J. Radiat. Oncol. Biol. Phys. 77, 1066–1071 (2010).
Good, J. S. & Harrington, K. J. The hallmarks of cancer and the radiation oncologist: updating the 5Rs of radiobiology. Clin. Oncol. (R. Coll. Radiol) 25, 569–577 (2013).
Hudis, C. A. et al. Proposal for standardized definitions for efficacy end points in adjuvant breast cancer trials: the STEEP system. J. Clin. Oncol. 25, 2127–2132 (2007).
Hanna, T. P., Shafiq, J., Delaney, G. P. & Barton, M. B. The population benefit of radiotherapy for cervical cancer: local control and survival estimates for optimally utilized radiotherapy and chemoradiation. Radiother. Oncol. 114, 389–394 (2015).
Mayr, N. A. et al. Longitudinal changes in tumor perfusion pattern during the radiation therapy course and its clinical impact in cervical cancer. Int. J. Radiat. Oncol. Biol. Phys. 77, 502–508 (2010).
Gadducci, A. et al. Pattern of failures and clinical outcome of patients with locally advanced cervical cancer treated with a tailored integrated therapeutic approach. Anticancer Res. 30, 3731–3735 (2010).
Rouzier, R. et al. Survival in cervix cancer patients treated with radiotherapy followed by radical surgery. Eur. J. Surg. Oncol. 31, 424–433 (2005).
Kawaguchi, R., Furukawa, N., Kobayashi, H. & Asakawa, I. Posttreatment cut-off levels of squamous cell carcinoma antigen as a prognostic factor in patients with locally advanced cervical cancer treated with radiotherapy. J. Gynecol. Oncol. 24, 313–320 (2013).
D'Amico, A. V. et al. Surrogate endpoints for prostate cancer-specific mortality after radiotherapy and androgen suppression therapy in men with localised or locally advanced prostate cancer: an analysis of two randomised trials. Lancet Oncol. 13, 189–195 (2012).
Ray, M. E. et al. Potential surrogate endpoints for prostate cancer survival: analysis of a phase III randomized trial. J. Natl Cancer Inst. 101, 228–236 (2009).
Machtay, M. et al. Defining local-regional control and its importance in locally advanced non-small cell lung carcinoma. J. Thorac. Oncol. 7 716–722 (2012).
Mauguen, A. et al. Surrogate endpoints for overall survival in chemotherapy and radiotherapy trials in operable and locally advanced lung cancer: a re-analysis of meta-analyses of individual patients' data. Lancet Oncol. 14, 619–626 (2013).
Machtay, M. et al. Prediction of survival by [18F]fluorodeoxyglucose positron emission tomography in patients with locally advanced non-small-cell lung cancer undergoing definitive chemoradiation therapy: results of the ACRIN 6668/RTOG 0235 trial. J. Clin. Oncol. 31, 3823–3830 (2013).
Hiltermann, T. J. et al. Circulating tumor cells in small-cell lung cancer: a predictive and prognostic factor. Ann. Oncol. 23, 2937–2942 (2012).
Milosevic, M. et al. Radiotherapy for bladder cancer. Urology 69, 80–92 (2007).
Feuerstein, M. A. & Goenka, A. Quality of life outcomes for bladder cancer patients undergoing bladder preservation with radiotherapy. Curr. Urol. Rep. 16, 75 (2015).
Rödel, C. et al. Combined-modality treatment and selective organ preservation in invasive bladder cancer: long-term results. J. Clin. Oncol. 20, 3061–3071 (2002).
Huddart, R. A. et al. Randomized noninferiority trial of reduced high-dose volume versus standard volume radiation therapy for muscle-invasive bladder cancer: results of the BC2001 trial (CRUK/01/004). Int. J. Radiat. Oncol. Biol. Phys. 87, 261–269 (2013).
Sjoquist, K. M. et al. Survival after neoadjuvant chemotherapy or chemoradiotherapy for resectable oesophageal carcinoma: an updated meta-analysis. Lancet Oncol. 12, 681–692 (2011).
Berger, A. C. et al. Complete response to neoadjuvant chemoradiotherapy in esophageal carcinoma is associated with significantly improved survival. J. Clin. Oncol. 23, 4330–4337 (2005).
Swisher, S. G. et al. 2-Fluoro-2-deoxy-D-glucose positron emission tomography imaging is predictive of pathologic response and survival after preoperative chemoradiation in patients with esophageal carcinoma. Cancer 101, 1776–1785 (2004).
Patel, U. B. et al. Magnetic resonance imaging-detected tumor response for locally advanced rectal cancer predicts survival outcomes: MERCURY experience. J. Clin. Oncol. 29, 3753–3760 (2011).
Glynne-Jones, R., Adams, R. A., Jitlal, M. & Meadows, H. End points in anal cancer: hopes for a common language. J. Clin. Oncol. 32, 1281–1282 (2014).
Day, F. L. et al. FDG-PET metabolic response predicts outcomes in anal cancer managed with chemoradiotherapy. Br. J. Cancer 105, 498–504 (2011).
Schwarz, J. K. et al. Tumor response and survival predicted by post-therapy FDG-PET/CT in anal cancer. Int. J. Radiat. Oncol. Biol. Phys. 71, 180–186 (2008).
Deniaud-Alexandre, E. et al. Results of definitive irradiation in a series of 305 epidermoid carcinomas of the anal canal. Int. J. Radiat. Oncol. Biol. Phys. 56, 1259–1273 (2003).
Tzeng, C. W. et al. Serum carbohydrate antigen 19-9 represents a marker of response to neoadjuvant therapy in patients with borderline resectable pancreatic cancer. HPB (Oxford) 16, 430–438 (2014).
Rudra, S. et al. Evaluation of predictive variables in locally advanced pancreatic adenocarcinoma patients receiving definitive chemoradiation. Pract. Radiat. Oncol. 2, 77–85 (2012).
Amin, A. et al. Pentavalent technetium-99m-dimercaptosuccinic acid [Tc-99m (V) DMSA] brain SPECT: does it have a place in predicting survival in patients with glioblastoma multiforme? J. Neurooncol. 121, 303–309 (2015).
Shah, G. D. et al. Comparison of linear and volumetric criteria in assessing tumor response in adult high-grade gliomas. Neuro Oncol. 8, 38–46 (2006).
Chen, Y.-P. et al. Potential surrogate endpoints for overall survival in locoregionally advanced nasopharyngeal carcinoma: an analysis of a phase III randomized trial. Sci. Rep. 5, 12502 (2015).
Michiels, S. et al. Surrogate endpoints for overall survival in locally advanced head and neck cancer: meta-analyses of individual patient data. Lancet Oncol. 10, 341–350 (2009).
Xie, P. et al. 18F-FDG PET or PET-CT to evaluate prognosis for head and neck cancer: a meta-analysis. J. Cancer Res. Clin. Oncol. 137, 1085–1093 (2011).
Hentschel, M. et al. Early FDG PET at 10 or 20 Gy under chemoradiotherapy is prognostic for locoregional control and overall survival in patients with head and neck cancer. Eur. J. Nucl. Med. Mol. Imaging 38, 1203–1211 (2011).
Cortazar, P. et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet 384, 164–172 (2014).
Author information
Authors and Affiliations
Consortia
Contributions
All the authors substantially contributed to discussing the content, writing, review and editing of manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
R.A.S. has received research funding from Sirtex Technology and consultation fees from Affidea, BTG plc, Cancer Research Technology, Sirtex Medical, and Vertex. O.A. is an employee of Eisai. S.K. and S.B. are employees of Pfizer Ltd. L.B. has received honorarium from Eli Lilly, Pfizer and Roche. G.C. is an employee and shareholder of AstraZeneca. A.N.C. is an employee and stockholder of MISSION therapeutics. L.D. is an employee of Merck KGaA. R.F. is an employee of Astex Pharmaceuticals. J.G. is an employee of Vertex Pharmaceuticals (Europe) Ltd and owns shares in Vertex Pharmaceuticals Inc. R.M.H. is an employee of BTG International Ltd, a manufacturer of specialist healthcare products. A.R.H. is an employee and stock holder of Eli Lilly. T.I. receives research support from AstraZeneca, Medimmune and Roche. T.M. is an employee of Johnson & Johnson. J.R.P. is an employee of, and holds stock in, Vertex Pharmaceuticals. M.S. is an employee of Genentech and stockholder of Roche. I.J.S. is a recipient of a research grant from AstraZeneca. S.R.W. receives research funding from Astex Pharmaceuticals and AstraZeneca. R.P., T.A.G., J.K.S., R.C., R.A.A., R.D.B., S.R.B., H.B., A.J.C., M.D.F., E.H., K.J.H., T.H., M.A.H., H.J., A.S.K., F.M., J.P.B.O'C., M.P.S., D.S-M. and J.S. declare no competing interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sharma, R., Plummer, R., Stock, J. et al. Clinical development of new drug–radiotherapy combinations. Nat Rev Clin Oncol 13, 627–642 (2016). https://doi.org/10.1038/nrclinonc.2016.79
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrclinonc.2016.79
This article is cited by
-
Results and lessons learnt from the WISTERIA phase I trial combining AZD1775 with cisplatin pre- or post-operatively in head and neck cancer
BJC Reports (2024)
-
CHARIOT: a phase I study of berzosertib with chemoradiotherapy in oesophageal and other solid cancers using time to event continual reassessment method
British Journal of Cancer (2024)
-
Engineering biomimetic nanosystem targeting multiple tumor radioresistance hallmarks for enhanced radiotherapy
Science China Life Sciences (2024)
-
Deep Insight of Design, Mechanism, and Cancer Theranostic Strategy of Nanozymes
Nano-Micro Letters (2024)
-
Cancer-associated fibroblasts in radiotherapy: Bystanders or protagonists?
Cell Communication and Signaling (2023)
