
Abstract
Structural, biochemical and biophysical studies of eukaryotic membrane proteins are often hampered by difficulties in overexpression of the candidate molecule. Baculovirus transduction of mammalian cells (BacMam), although a powerful method to heterologously express membrane proteins, can be cumbersome for screening and expression of multiple constructs. We therefore developed plasmid Eric Gouaux (pEG) BacMam, a vector optimized for use in screening assays, as well as for efficient production of baculovirus and robust expression of the target protein. In this protocol, we show how to use small-scale transient transfection and fluorescence-detection size-exclusion chromatography (FSEC) experiments using a GFP-His8–tagged candidate protein to screen for monodispersity and expression level. Once promising candidates are identified, we describe how to generate baculovirus, transduce HEK293S GnTI− (N-acetylglucosaminyltransferase I-negative) cells in suspension culture and overexpress the candidate protein. We have used these methods to prepare pure samples of chicken acid-sensing ion channel 1a (cASIC1) and Caenorhabditis elegans glutamate-gated chloride channel (GluCl) for X-ray crystallography, demonstrating how to rapidly and efficiently screen hundreds of constructs and accomplish large-scale expression in 4–6 weeks.
Similar content being viewed by others
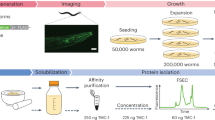


Introduction
Since the initial observation that insertion of a human cytomegalovirus (CMV) promoter or a Rous sarcoma virus (RSV) promoter into an Autographa californica multiple nucleopolyhedrosis virus (AcMNPV; from here on referred to as baculovirus) transfer vector allowed for the expression of foreign genes in hepatocytes and other mammalian cell lines1,2, BacMam has been used for a growing number of applications. These applications include drug discovery (identification and development of new therapeutic agents) through recombinant protein expression for cell-based functional assays using G protein–coupled receptors (GPCRs)3,4, nuclear receptors5, ion channels6,7 and ATP-binding cassette drug transporters8. More recently, BacMam has been used for large-scale protein production for crystallography9,10,11,12,13,14,15,16,17,18,19,20. The success of these applications, however, depends in part on the efficient production and amplification of baculovirus and on subsequent large-scale transduction and heterologous protein expression. In addition to these challenges, obtaining sufficient quantities of membrane protein for crystallography is frequently compounded by low levels of expression and instability of the candidate membrane protein, thus requiring screening of many constructs. Furthermore, some mammalian membrane proteins require specific post-translational modifications and a near-native lipid environment, thus rendering expression in insect cells or in yeast untenable. Taken together, these complexities can result in a high cost for heterologous membrane protein expression in mammalian cells, and thus improving the efficiency of the process is important.
Here we describe methods for screening constructs and optimizing heterologous expression of membrane proteins from BacMam-transduced HEK293S GnTI− (N-acetylglucosaminyltransferase I–negative) cells for purification and crystallization (Fig. 1). We have constructed a vector (pEG BacMam) for high-level expression in mammalian cells using elements derived from a previously described vector, pVLAD10. Once genes of candidate membrane proteins are fused in frame with a GFP tag and cloned into pEG BacMam, they can be rapidly screened for expression and monodispersity using transient transfection in adherent cells coupled with FSEC21,22. We also optimized virus amplification and protein expression protocols such that the cost and time for expressing most of the membrane proteins in HEK293 GnTI− cells are similar to or better than those of expression in Sf9 cells.

After one or more rounds of screening, a few potential candidates are chosen for large-scale protein expression.
This protocol is exemplified using two proteins expressed in mammalian cells: Gallus gallus cASIC1 (refs. 16,23) and C. elegans GluCl24,25. This protocol is now in standard use in our laboratory for mammalian-expressed membrane proteins15,16,17,20.
Development of the protocol
To increase the heterologous expression of challenging membrane proteins, we first constructed pEG BacMam for high-level protein expression in mammalian cells with the ability to express multiprotein complexes from a single vector (Fig. 2). To do this, we chemically synthesized genetic elements derived from the previously described BacMam vector pVLAD10, which include a strong CMV promoter for robust transcription, a synthetic intron for efficient RNA splicing and mRNA processing, and a WPRE motif for efficient mRNA processing, stability and export. These chemically synthesized elements were combined with the pFBDM (ref. 26), a bicistronic vector with a restriction enzyme module that allows the assembly of multiple expression cassettes, to generate pEG BacMam. After the gene of interest is cloned into pEG BacMam, we screen constructs by small-scale transfection/FSEC before moving to the time-consuming process of virus amplification21.

For expression in mammalian cells, genes of interest are cloned into the multiple cloning site behind the CMV promoter using unique restriction sites. Elements that are important for high-level expression are shown, including those that are important for transcription initiation (CMV promoter), transcription termination (SV40 poly-A late signal) and mRNA processing (intron and WPRE motif). Also indicated are the elements that are important for insect cell expression and baculovirus amplification, including promoters (polh and p10), terminators (SV40 and HSV-tk), transposon elements (Tn7L and Tn7R) and resistance markers (ampicillin and gentamicin). IE, immediate/early.
In our research to optimize protein expression, we compared the expression of cASIC1 and GluCl in mammalian cells and insect cells. We show that fivefold more GluCl pentamer can be obtained in mammalian cells. In the case of cASIC1, not only can twofold more trimer be obtained in mammalian cells but also the protein is more monodisperse and experiences less spontaneous cleavage of the GFP-His8 tag.
Although other HEK cell lines can be used, for screening and expression we typically use HEK293S GnTI− cells, a mammalian cell line that expresses proteins that are more mannose-rich and are thus easily removed using endoglycosidases such as EndoH (ref. 27). Although the use of these cells and EndoH can reduce the heterogeneity caused by complex glycans that can create problems in crystallographic studies, it may not be beneficial for every protein. Therefore, it is advantageous to test protein expression in other mammalian cell lines, as well as to determine whether the use of EndoH affects the solubility and the heterogeneity of the glycoprotein.
To determine the expression level and monodispersity of the candidate membrane protein, HEK293S GnTI− cells are transfected with the pEG BacMam plasmid containing the gene of interest; collected after 48 h; and then solubilized in a buffer containing n-dodecyl-β-D-maltoside (C12M), maltose-neopentyl glycol28 (MNG-3), or other detergent. The resulting supernatant is analyzed by FSEC (Fig. 3). As shown in Figure 3, removing the 64 residues from the C terminus of cASIC1 (cASIC1 Δ463) increases monodispersity and reduces cleavage of the GFP-His8 tag. In addition to removing flexible termini, there are many methods that can be used to optimize the expression and stability of proteins, including codon optimization, surface entropy reduction and thermostability mutations29,30,31. Small-scale transfection followed by whole-cell solubilization and FSEC allows the screening of hundreds of candidates in ≤1 month.

Cells were collected 48 h after transfection, and solubilized extracts were analyzed by FSEC to determine the behavior of the fusion proteins. c.p.s., counts per second.
Once a promising candidate is identified, the plasmid is transformed into the DH10Bac Escherichia coli strain to generate the recombinant bacmid DNA, which is then used to transfect insect cells to generate BacMam virus. We have detailed our methods for isolation of bacmid DNA, transfection of Sf9 cells and baculovirus amplification, which we use to reduce costs and ensure good-quality BacMam virus. We have found for some constructs that the multiplicity of infection (MOI) during virus amplification is 10–100-fold below the range recommended by the Bac-to-Bac system (Invitrogen) protocol (http://www.lifetechnologies.com/us/en/home/life-science/protein-expression-and-analysis/protein-expression/insect-expression/bac-to-bac-baculovirus-expression-system.html). We have also found that a low MOI (MOI of 2 or less) is sufficient for mammalian cell transduction and that too much virus results in low cell numbers, possibly owing to too much Sf9 medium or virus added to the culture. Therefore, before virus amplification or transduction of mammalian cells for protein expression, virus titer should be determined using the endpoint dilution assay32, flow cytometric assay33,34 or the viral plaque assay35.
In addition to MOI, we also explored different growth and expression conditions for BacMam-transduced HEK293S GnTI− cells to boost protein expression. After testing several types of media for the growth of suspension cells, we found that the use of Gibco FreeStyle 293 expression medium (Invitrogen) allowed for increased growth rates and reduced cell clumping of HEK293S GnTI− cells in suspension. To further minimize cell clumping, we also assessed the growth of suspension cells in different vessels, including square bottles, flat-bottom flasks and baffled Erlenmeyer flasks. We found that baffled flasks minimized cell clumping and promoted cell growth. To reduce costs, the polycarbonate Erlenmeyer flasks can be washed, autoclaved and used again up to 20 times. If after autoclaving the filter in the cap deteriorates, a replacement cap can be purchased.
Previously, it has been shown that lowering the temperature after transduction or transfection enhances protein expression in mammalian cells36,37,38,39,40,41,42,43. In a time-course experiment for cASIC1, we found that at the optimal collection time of 72 h after transduction, there was a 4.3-fold increase in expression at 30 °C compared with that at 37 °C (Fig. 4a). Gaussian fitting, as described previously21,22, was performed on the FSEC profiles to determine the peak area of cASIC1 trimer. A graph of the trimer peak area shows that the expression of cASIC1 is higher at 30 °C than at 37 °C for most time points (Fig. 4b). To monitor the expression of GluCl in HEK293S GnTI− cells, an EGFP GluCl gene fusion25,44 was cloned into pEG BacMam and used to generate BacMam virus. At the optimal collection time of 72 h after transduction for cells expressing GluCl, there was a 9.5-fold increase in expression at 30 °C with higher expression of GluCl at all time points at 30 °C (Fig. 4c,d). To show that the proteins present in the major peaks for cASIC1 and GluCl are of the expected molecular weight, peak fractions were collected and analyzed by SDS-PAGE, followed by western blot analysis using an antibody against GFP (Fig. 4e). In fact, for the expression of most proteins in HEK293S GnTI− cells, we have found that lowering the temperature during expression increases protein yields at least twofold (A.G., C.-H.L., K.H.W., J.C.M., D.P.C. and E.G., unpublished data). In some cases, lowering the temperature is essential in order to obtain monodisperse, well-folded protein.

(a–d) HEK293S GnTI− cells (3 × 106 cells/ml) were transduced with BacMam virus for cASIC1 Δ463 (a,b) or GluCl-EGFP (c,d) at an MOI of 1 and incubated at 37 °C. After 8 h, 10 mM sodium butyrate was added and cultures were left at 37 °C or shifted to 30 °C. Time points were taken at the indicated times, samples were frozen and later solubilized with 40 mM C12M in TBS (pH 8) and analyzed by FSEC. (a,c) Representative FSEC profiles from cASIC1 Δ463 (a) and GluCl-EGFP (c) detected by GFP fluorescence. (b,d) Comparison of expression at 37 °C and 30 °C from cASIC1 Δ463 (b) and GluCl-EGFP (d) on the basis of the peak area estimated by Gaussian fitting. (e) Western blot analysis using an anti-GFP polyclonal antibody of peak fractions isolated from FSEC profiles from cASIC1 Δ463 (a) and GluCl-EGFP (c). a.u., arbitrary units; c.p.s., counts per second.
Finally, the use of histone deacetylase inhibitors has been shown to enhance protein expression in HEK293S cells (refs. 10,45). We found that for most membrane proteins histone deacetylase inhibitors boost expression. For cASIC1, there is a sevenfold increase in expression at 72 h after transduction, as well as higher expression of cASIC1 at all time points at which HEK293S GnTI− cells are treated with 10 mM sodium butyrate (Fig. 5a,b). Previously published data suggest that valproic acid is more efficient than sodium butyrate at enhancing recombinant protein production from mammalian cells45. However, we find that both sodium butyrate and valproic acid enhance protein expression from BacMam-transduced HEK293S GnTI− cells (Fig. 5c). Typically, sodium butyrate is added to cultures between 8 and 24 h after transduction; however, the amount and time of sodium butyrate addition should be optimized for each protein. Overall optimization of conditions (construct, MOI, cell density, temperature and collection time of BacMam-transduced HEK293S GnTI− cells) could either increase expression or decrease aggregation, leading to more properly folded protein, and therefore the most favorable conditions for each protein should be determined before attempting a large-scale expression.

(a,b) Comparison of cASIC1 expression with and without sodium butyrate; representative FSEC profiles from cASIC1 Δ463 detected by GFP fluorescence (a) and comparison of expression from cASIC1 Δ463 on the basis of the peak area estimated by Gaussian fitting (b) in the presence or absence of sodium butyrate. (c) Representative FSEC profiles from GluCl-EGFP in the presence of sodium butyrate or valproic acid. c.p.s., counts per second.
Other applications of the method
The protocol described here could also be used to optimize the expression of heteromers or protein complexes in mammalian cells. One option to simultaneously express multiple proteins is to co-infect with multiple BacMam viruses (with an optimized MOI for each virus). Alternatively, pEG BacMam could be used to express multiprotein complexes by combining two pEG BacMam plasmids using unique restriction enzyme sites. As a result, multiple genes could be transduced by a single BacMam virus, allowing for the simultaneous expression of two or more genes.
Some variants of pEG BacMam also contain vesicular stomatitis Indiana virus glycoprotein (VSIV-G) under control of the P10 promoter (a baculovirus-specific promoter). VSIV-G is a viral capsule protein important for mediating viral entry, and it has been shown to increase the transduction efficiency of baculovirus for some mammalian cells46. The P10 promoter could be used to drive the expression of VSIV-G in insect cells, allowing the incorporation of VSIV-G into the baculovirus to enhance transduction from other mammalian cell lines, such as the human lung carcinoma line A549 and the human hepatoma lines HepG2 and HuH7 (ref. 46).
Comparison with other methods
Many methods have been used for the overexpression of mammalian proteins, including plasmid transfection, stable cell lines and viral expression systems such as Sindbis virus, vaccinia virus and Semliki Forest virus (SFV), as well as AcMNPV47. Compared with BacMam, each of these methods has advantages and disadvantages in terms of cost, time, efficiency, safety and reproducibility.
One such transient expression method involves transfection of plasmid DNA into adherent cells or cultures, and overexpression is either immediate or induced, depending on the promoter. Plasmid transfection is fast, safe and easy to use for high-throughput screening48,49,50. However, if commercial transfection reagents and plasmid isolation kits are used, plasmid transfection can be expensive for large-scale expression. Furthermore, the level of expression using plasmid transfection can be limited by the plasmid size, the number of plasmids transfected and cytotoxic effects that have been observed with many transfection reagents. The use of BacMam can be cheaper than plasmid transfection for large-scale expression and multiple rounds of expression. In addition, BacMam is not limited by the gene number or size. In our hands, the level of protein expression, especially multisubunit proteins, is higher with BacMam than with plasmid transfection.
Stable expression in mammalian cells requires the integration of a transfected transgene into the cell's genome using Geneticin or other selection methods. Once clonal cell lines are generated and sorted for high-level producers, long-term overexpression from stably transfected cells can be robust, easy and consistent51,52. Furthermore, stable cell lines can be generated using regulated expression, such as the tetracycline-inducible expression system, thus allowing for large-scale expression of proteins that are cytotoxic to the cell51,53. Although stable expression enables the production of large quantities of protein, when compared to BacMam generating a stable cell line is time consuming.
Although other viral expression systems for protein expression such as lentiviruses54, adenoviruses55, Sindbis virus56,57, vaccinia virus58 and SFV59,60 exist, SFV has been used to express a large number of membrane proteins consisting of mostly GPCRs47. To make virus, candidate genes are cloned into SFV plasmid and used as template for RNA synthesis. The RNA is then co-transfected (either using electroporation or liposome reagents) with helper RNA and packaged into SFV particles that can then be used to infect cells in culture. Although SFV can be easily used for small-scale studies by transfecting synthesized RNA into cells and by analyzing expression, using SFV for large cultures is more challenging than BacMam owing to the amount of RNA that is needed to make virus for large-scale studies.
An effective method using BacMam was recently described to produce milligram quantities of proteins sufficient for crystallization10. However, the methods outlined therein to produce recombinant BacMam virus using the BD BaculoGold system are not as cost-efficient as the Bac-to-Bac method if multiple constructs are expected to be generated. In addition, we optimized growth and expression conditions using BacMam in order to express sufficient amounts of our desired membrane proteins.
Another commonly used method for the overexpression of proteins is AcMNPV baculovirus infection of insect cells. Although insect cells such as Sf9 cells have been used in our laboratory previously to provide sufficient protein for crystallization studies25,61,62,63,64, some membrane proteins require a near-native environment to help ensure functional expression. The advantages for the expression of eukaryotic membrane proteins in mammalian cells over Sf9 include improved post-translational modifications such as N-linked glycosylation65,66 and a different lipid environment that contains higher amounts of cholesterol. We performed a side-by-side comparison of protein expression from insect cells and mammalian cells. We found a twofold increase in cASIC1 trimer and an increase in homogeneity (Fig. 6a,b), and we found that fivefold more GluCl pentamer can be obtained in mammalian cells (Fig. 6c,d).

(a–d) HEK293S GnTI− cells were transduced with BacMam virus for cASIC1 Δ463 (a,b) or GluCl-EGFP (c,d), as described above. Sf9 cells (3 × 106 cells per ml) were infected with baculovirus cASIC1 Δ463 (a,b) or GluCl-EGFP (c,d) and placed at 27 °C. After 18 h, Sf9 cultures were left at 27 °C or shifted to 20 °C. Time points were taken, frozen and solubilized as described above. (a,c) Representative FSEC profiles from cASIC1 Δ463 (a) and GluCl-EGFP (c) detected by GFP fluorescence. (b,d) Comparison of expression from cASIC1 Δ463 (b) and GluCl-EGFP (d) on the basis of the peak area estimated by Gaussian fitting. a.u., arbitrary units; c.p.s., counts per second.
Materials
REAGENTS
-
pEG BacMam plasmid containing the gene of interest in frame with a GFP tag and a tag appropriate for affinity chromatography (such as a His8 tag) cloned downstream of the CMV promoter (Fig. 2)
-
HEK293S GnTI− cells (ATCC, cat. no. CRL-3022)
-
Gibco FreeStyle 293 expression medium (Life Technologies, cat. no. 12338-018)
-
US certified γ-irradiated FBS (Life Technologies, cat. no. 0984018DJ)
-
DMEM (with 4.5 g per liter glucose, L-glutamine and sodium pyruvate; Corning/Cellgro, cat. no. 10-013)
-
Opti-MEM I reduced serum medium (Life Technologies, cat. no. 31985-088)
-
Lipofectamine 2000 (Life Technologies, cat. no. 11668-027)
-
PBS (without calcium and magnesium; Corning/Cellgro, cat. no. 21-040-CM)
-
Trypsin-EDTA (Corning/Cellgro, cat. no. 25-052-CV)
-
Tris base (Fisher Scientific, cat. no. BP152)
-
NaCl (Sigma-Aldrich, cat. no. 59888)
-
C12M (Affymetrix, cat. no. D310)
-
PMSF (Sigma-Aldrich, cat. no. 78830)
-
Leupeptin (Sigma-Aldrich, cat. no. L0649)
-
Aprotinin (Sigma-Aldrich, cat. no. A1153)
-
Pepstatin (Sigma-Aldrich, cat. no. P4265)
-
Sodium butyrate (Sigma-Aldrich, cat. no. 303410)
-
Valproic acid (Sigma-Aldrich, cat. no. P4543)
-
DH10Bac competent cells (Life Technologies, cat. no. 10361-012)
-
Sf9 Easy Titer cell line32
-
Cellfectin II reagent (Life Technologies, cat. no. 10362-100)
-
Sf9 cells (Life Technologies, cat. no. 12659017)
-
Sf-900 III serum-free medium (SFM) (Life Technologies, cat. no. 12658-027)
-
QIAprep spin miniprep kit (Qiagen, cat. no. 27104)
-
HyClone SFX-Insect medium (GE/Hyclone, cat. no. SH30278.02)
-
G 418 disulfate salt (Sigma-Aldrich, cat. no. A1720-1g)
-
Trypan blue solution (Corning/Cellgro, cat. no. 25-900-CL)
EQUIPMENT
-
Incubator (Thermo Scientific, cat. no. 3950)
-
Tissue culture plate (100 mm; BD Falcon, cat. no. 35300)
-
Tissue culture plate (six well; BD Falcon, cat. no. 353046)
-
Erlenmeyer baffled flasks (2,000 ml; BioExpress, cat. no. F-5909-2000B)
-
Nunc EasYFlask (75 cm2, Filter Cap; Thermo Scientific, cat. no. 156499)
-
Syringe filters (polyethersulfone (PES) 13 mm diameter, 0.22 μm, polypropylene (PP) housing; Argos, cat. no. FE12S)
-
Filter systems (250 ml, 0.22 μm; Corning, cat. no. 430767)
-
Virus Counter 2100 (ViroCyt)
-
Optima TL ultracentrifuge (Beckman)
-
Fluorescence-detection size-exclusion chromatography system (FSEC; Equipment Setup)21
-
Tissue culture plate (Costar 96-well black clear-bottom plates; Costar/Corning, cat. no. 3603)
-
Sterile disposable reagent reservoirs (50 ml white; Costar/Corning, cat. no. 4870)
-
Cluster tube system eight-tube strip (Costar/Corning product, cat. no. 4413)
-
Cell scrapers (handle 18 cm blade 1.8 cm; Corning, cat. no. 353085)
-
Hemocytometer (Hausser Scientific, cat. no. 1492)
-
Shimadzu fluorometer (RF-20A)
REAGENT SETUP
DMEM medium
-
To 500 ml of DMEM medium, add 50 ml of FBS. Store the medium at 4 °C for 1 month.
Suspension medium
-
To 1 liter of FreeStyle 293 Expression medium, add 20 ml of FBS. Store the medium at 4 °C for 1 month.
Suspension medium
-
To 1 liter of HyClone SFX-Insect medium, add 50 ml of FBS and 150 μg/ml G418. Store the medium at 4 °C for 1 month.
Sodium butyrate (2 M)
-
Dissolve 11 g of sodium butyrate with water to a final volume of 50 ml, and filter-sterilize using a 0.2-μm filter inside a biological safety cabinet. Store it at −20 °C for at least 1 month.
Tris-buffered saline (TBS)
-
Mix 20 mM Tris-HCl (pH 8) and 150 mM NaCl. Store TBS at room temperature (RT; 25 °C) for at least 1 month.
Solubilization buffer
-
Mix 20 mM Tris-HCl (pH 8), 150 mM NaCl and 40 mM C12M. Chill the buffer to 4 °C. Immediately before use, add 1 mM PMSF, 200 μM aprotinin, 2 μg/ml leupeptin and 2 μM pepstatin A. Discard any unused buffer.
FSEC buffer
-
Mix 20 mM Tris HCl (pH 8), 150 mM NaCl and 1 mM C12M. Filter the buffer using a 0.2-μm filter. Store the buffer at 4 °C for up to 1 week.
Purification of plasmid DNA
-
Purify plasmid DNA using the QIAprep spin miniprep kit (Qiagen) or another suitable method.
Growth and maintenance of adherent HEK293S GnTI− cells
-
Cells are cultured as previously described51.
Growth and maintenance of suspension HEK293S GnTI− cells
-
HEK293S GnTI− cells are maintained as described in Box 1.
Growth and maintenance of Sf9 cells
-
Sf9 cells are maintained as suspension cultures at 27 °C in Sf-900 III SFM medium. Isolation of bacmid DNA, transfection of Sf9 cells and amplification of virus are methods modified from the Bac-to-Bac system (Invitrogen), which we use to reduce costs and ensure production of good-quality BacMam virus.
Growth and maintenance of Sf9 Easy Titer cell line
-
Sf9 Easy Titer cell lines are maintained as adherent cells at 27 °C in HyClone SFX-Insect medium, as described in Box 2.
BacMam virus titer determination
-
Determine the titer of the BacMam virus using one of the several methods for virus titer determination. We prefer using the Sf9 Easy Titer cell line and the endpoint dilution assay32 (Box 2) or the flow cytometric assay33,34.
EQUIPMENT SETUP
FSEC
-
In our laboratory, this is performed as described by Kawate and Gouaux21. The analyte is loaded onto a Superose 6 column (10/30, Amersham Biosciences) that has been pre-equilibrated with FSEC buffer. Separation is performed at a flow rate of 0.5 ml/min. The eluent from the SEC column is passed through a Shimadzu fluorometer (RF-20A) fitted with a flow cell, as described in the manufacturer's instructions. The fluorometer settings are as follows: band-pass, 3 nm/3 nm; excitation, 488 nm; emission, 507 nm; time increment, 1 s; integration time, 1 s; and recording time, 3,000–3,600 s. Calibration with known quantities of GFP have demonstrated that 1–10 ng of GFP can readily be detected.
Procedure
Cell seeding (day 1)
Timing 15 min
-
1
Add 1 × 106 HEK293S GnTI− cells in 2 ml of DMEM supplemented with FBS to each well of a six-well culture plate. Incubate the cells at 37 °C with 5% CO2 for 16–24 h.
Caution
Cell cultures are a potential biohazard. Work in an approved laminar flow hood using aseptic techniques, and check institutional and governmental guidelines for recommended protective clothing and proper disposal of waste before performing experiments.
Small-scale transient transfection to screen constructs (day 2)
Timing 45 min
-
2
For each well, prepare an autoclaved 1.5-ml centrifuge tube. By using a pipette, add 4 μl of Lipofectamine 2000 into 50 μl of Opti-MEM I.
-
3
Add 1 μg of Qiagen miniprep–purified DNA into 50 μl of Opti-MEM I in a separate 1.5-ml centrifuge tube.
-
4
Add DNA/Opti-MEM I mixture to the Opti-MEM/Lipofectamine mixture; gently mix and incubate it for 20 min at RT.
-
5
Pipette the Opti-MEM I–DNA mixture dropwise onto 70–80% confluent HEK293S GnTI− cells. Ensure even dispersal.
-
6
After 8–24 h, replace the medium with DMEM plus 10 mM sodium butyrate.
-
7
Incubate the cells at 37 °C with 5% CO2 for 2 d.
Screening the constructs by FSEC for monodispersity and expression level (day 4)
Timing 3 h
-
8
Aspirate off the medium and wash the transfected adherent cells carefully with 2 ml of TBS.
-
9
Add 1 ml of TBS to each well, collect the cells and transfer them to a 1.5-ml centrifuge tube.
-
10
Centrifuge the cells at 1,500g for 5 min at 4 °C.
-
11
Remove the supernatant and resuspend the cell pellet in 200 μl of solubilization buffer.
-
12
Nutate the samples for 1 h at 4 °C.
-
13
Centrifuge the solubilized sample at 70,000g in a TL100 ultracentrifuge for 40 min at 4 °C.
-
14
Collect the supernatant and analyze 50 μl by FSEC21. Allow 1 h for each sample to be analyzed by FSEC. Samples should be stored at 4 °C until analysis.
-
15
Identify the best-expressed and monodisperse candidate via FSEC (Fig. 3 and Kawate and Gouaux21).
Transformation of DH10Bac E. coli (day 5)
Timing 1 h
-
16
Transform purified plasmid DNA into DH10Bac E. coli for transposition into the bacmid, as described in the Bac-to-Bac system protocol (Invitrogen; http://www.lifetechnologies.com/us/en/home/life-science/protein-expression-and-analysis/protein-expression/insect-expression/bac-to-bac-baculovirus-expression-system.html).
Inoculation of bacmid-containing cultures (day 7)
Timing 15 min
-
17
Inoculate 5 ml of LB medium containing 50 μg/ml kanamycin, 7 μg/ml gentamicin and 10 μg/ml tetracycline with a white colony, and allow the cells to grow overnight at 37 °C.
Isolation of bacmid (day 8)
Timing 1 h
-
18
Centrifuge the cells for 10 min at 1,500g at RT.
-
19
(Optional) Make a glycerol stock of the DH10Bac E. coli containing the bacmid DNA for future bacmid DNA isolation. In an autoclaved 1.5-ml centrifuge tube, take 250 μl of cell suspension (from Step 17) and add 250 μl of sterile 50% (vol/vol) glycerol; mix it and store it for years at −80 °C.
-
20
Discard the supernatant and resuspend the pellet in 200 μl of P1 (Qiagen kits). Transfer the suspension into a 1.5-ml centrifuge tube.
-
21
Add 200 μl of P2 (included in Qiagen kits) and mix by inverting the centrifuge tube a few times.
Caution
Do not vortex the samples, as this could shear the bacmid DNA.
-
22
Add 200 μl of N3 (included in Qiagen kits) and mix by inverting the centrifuge tube a few times; centrifuge the tube for 10 min at 1,500g at RT.
-
23
Transfer the supernatant to a 2-ml centrifuge tube, add 1 ml of isopropanol and gently invert the tube.
-
24
Place the tube for 10 min in a −20 °C freezer.
-
25
Centrifuge the tube at 1,500g for 15 min at RT.
-
26
Remove the supernatant, and preserve the pellet. Add 1 ml of 70% (vol/vol) ethanol and wash the pellet by gently inverting the centrifuge tube.
-
27
Centrifuge the tube at 1,500g for 15 min at RT.
-
28
Remove the supernatant and dry the pellet for 5 min.
-
29
Resuspend the pellet in 50 μl of autoclaved Milli-Q water. Determine the concentration of the bacmid DNA.
Caution
Do not pipette samples more than one or two times, as this could shear the bacmid DNA.
Pause point
Store the bacmid DNA at 4 °C until you are ready to proceed with Step 30 (up to 3 d).
Transfection of Sf9 cells with bacmid
Timing 2 h
-
30
Seed 9 × 105 of Sf9 cells in 2 ml of Sf-900 medium per well of a six-well plate.
-
31
Incubate the cells at 27 °C until they attach (∼20 min).
-
32
While waiting for the cells to attach, add 8 μl of Cellfectin II to 100 μl of Sf-900 III SFM medium in centrifuge tubes for each transfection.
-
33
In a different centrifuge tube, add 1 μg of bacmid DNA to 100 μl of Sf-900 III SFM.
-
34
Mix the Cellfectin II/Sf-900 III SFM media mixture and the bacmid DNA/Sf-900 III SFM mixture, and incubate for 30 min at RT.
-
35
Change the medium in each well with 2 ml of Sf-900 III SFM medium, and add the Cellfectin II/DNA mixture dropwise onto the Sf9 cells. Ensure even dispersal.
-
36
Incubate the cells for 72 h in a 27 °C incubator (make sure to have water inside the incubator to prevent strong evaporation of the medium).
-
37
Collect the supernatant containing P1 virus (∼2 ml from each well), and filter the medium containing P1 virus into a 2-ml centrifuge tube using a 3-ml syringe fitted with a small 0.2-μm filter. This is a stock of P1 virus that should be stored at 4 °C light protected for up to a month. Add 2% (vol/vol) FBS to the stabilized virus stock. It might also be helpful to use the titerless infected cells preservation and scale-up (TIPS) method67 to preserve Sf9 cells infected with P1 virus.
-
38
Determine the titer of the P1 BacMam virus using the Sf9 easy titer cell line and the end-point dilution assay or by using the Virus Counter 2100.
Infection of Sf9 cells with P1 virus to produce P2 virus
Timing 2 h
-
39
On the basis of the desired volume of P2 virus, add P1 virus to an MOI of 0.1 to 0.0001 to Sf9 cells that are 1.0–1.5 × 106 cells per ml in an Erlenmeyer flask of the corresponding size.
Critical Step
For the amplification of some viruses, we have found that it is essential to infect at a lower MOI than recommended by the Bac-to-Bac system (Invitrogen). Therefore, it may be important to determine the optimal MOI for the virus amplification before making a large amount of P2 virus.
-
40
Incubate the Sf9 cells infected with the P1 virus for 96 h in a 27 °C orbital shaker at 115 r.p.m.
Critical Step
For the amplification of some viruses, the collection time of the P2 BacMam virus may need to be optimized. We advise initially trying 72 and 96 h.
-
41
Centrifuge the cells at 8,000g for 15 min at 4 °C, and collect the supernatant containing P2 virus.
-
42
Filter the supernatant using disposable 0.2-μm filters (50-ml Steriflip filters from Millipore for small amounts or 250-ml, 0.5 l or 1-liter Corning filter systems for large amounts). Add 2% (vol/vol) FBS to the stabilized virus stock. This is a stock of P2 virus that should be stored at 4 °C, protected from light (we use aluminum foil) for up to 1 month.
-
43
Determine the titer of the P2 BacMam virus using the Sf9 Easy Titer cell line and the endpoint dilution assay or by using the Virus Counter 2100.
Transduction of suspension HEK293S GnTI− cells with BacMam virus
Timing 2 h
-
44
Expansion of HEK293S GnTI– cells should be prepared in advance (∼10 d in advance) so that a sufficient amount of cells are available on day 15. To expand HEK293S GnTI− cells, determine the total number of cells and percent viability using a hemocytometer and trypan blue exclusion, and make sure that the density of the cells are 2.5–3 × 106 cells per ml (from Box 1).
-
45
When a 25-ml culture of HEK293S GnTI− cells are 2.5–3 × 106 cells per ml, dilute the culture to 0.2 × 106 cells per ml in 200 ml and incubate the cells on an orbital shaker within a 37 °C incubator in the presence of 8% CO2 for ∼5 d until the density is 3 × 106 cells per ml.
-
46
On the basis of the volume of cells needed (2.4–6.4 liters), calculate the volume of medium that you need to add to dilute the culture to a seeding density of 0.2 × 106 cells per ml. We prefer to have a starting density of 0.2 × 106 cells per ml. For 2.4 liters, one will need 4.8 × 108 cells, ∼2.2 liters of medium and three 2-liter flasks.
-
47
Aseptically add the appropriate volume of prewarmed growth medium into the culture flask (the total volume should be 800 ml in a 2-liter flask). Split the culture to multiple flasks as needed, and incubate the cells on an orbital shaker within a 37 °C incubator in the presence of 8% CO2 for ∼5–6 d until the cells reach a density of 2–3.5 × 106 cells per ml.
-
48
Add BacMam P2 virus at an MOI of 1 to infect 2.4 liters of HEK293S GnTI− cells at a density of 2–3.5 × 106 cells per ml, and incubate the cells on an orbital shaker within a 37 °C incubator in the presence of 8% CO2.
Critical Step
The amount of virus added should not exceed >10% of the culture volume.
-
49
After 8–24 h at 37 °C, add 10 mM sodium butyrate and incubate the cells on an orbital shaker within a 30 °C incubator in the presence of 8% CO2.
-
50
Collect the cells 60–90 h after transduction by centrifugation for 20 min at 6,200g at 4 °C.
Critical Step
The collection time of BacMam-transduced HEK293S GnTI− cells should be determined before attempting a large-scale expression (Fig. 4).
Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
The entire protocol, starting from transfection (Step 1) to the collection of BacMam virus–transduced suspension HEK293S GnTI− cells (Step 50), takes ∼3 weeks to complete if a promising candidate is identified (Step 14). The hands-on timing for each stage of the PROCEDURE is summarized below.
Step 1, cell seeding: 15 min
Steps 2–7, small-scale transient transfection to screen constructs: 45 min
Steps 8–15, screening the constructs by FSEC for monodispersity and expression level: 3 h
Step 16, transformation of DH10Bac E. coli: 1 h
Step 17, inoculation of bacmid-containing cultures: 15 min
Steps 18–29, isolation of bacmid: 1 h
Steps 30–38, transfection of Sf9 cells with bacmid (producing P1 virus): 2 h (45 min for transfection + 15 min for P1 virus collection + 1 h for virus titer determination using the Virus Counter 2100)
Steps 39–43, infection of Sf9 cells with P1 virus to produce P2 virus: 2 h (15 min for infection of Sf9 cells with P1 virus + 45 min for collection of P2 virus + 1 h for virus titer determination using the Virus Counter 2100)
Steps 44–50, expansion of HEK293S GnTI− cells and transduction of suspension HEK293S GnTI− cells with BacMam virus: 2 h
Box 1, growth and maintenance of suspension HEK293S GnTI− cells: 15 min
Box 2, endpoint dilution assay: 1 h
Anticipated results
This protocol (as outlined in Fig. 1) has been used in our laboratory to successfully express cASIC1, Drosophila melanogaster dopamine transporter (DAT), N-methyl-D-aspartate (NMDA) receptors and many other membrane proteins in HEK293S GnTI− cells15,16,20,61. The time it takes to identify a promising candidate (Fig. 3) is likely to vary substantially depending on (for example) the number of flexible regions to be removed, surface entropy reduction mutations and thermostability mutations. Although few changes are made for cASIC1, several construct changes were needed for DAT and NMDA to obtain the best-expressed and monodisperse candidate via FSEC15,16,17,20. Once a promising candidate is identified, the most favorable conditions for MOI, cell density, expression time, temperature (Fig. 4) and the presence of histone deacetylase inhibitors (i.e., sodium butyrate; Fig. 5) should be determined for each protein before attempting a large-scale expression. The protocol for optimized expression can be completed in ∼3 weeks. Purification of membrane proteins from transduced HEK293S GnTI– cell pellets (which may include affinity chromatography, tag cleavage, removal of N-linked glycosylation and size-exclusion chromatography), depending on the candidate protein, can produce 0.25–1.5 mg of protein per liter of medium sufficient for crystallization. The protein expression and yield can vary depending on factors such as the titer of the virus, the toxicity of the protein when expressed and the stability of the protein.


References
Boyce, F.M. & Bucher, N.L. Baculovirus-mediated gene transfer into mammalian cells. Proc. Natl. Acad. Sci. USA 93, 2348–2352 (1996).
Hofmann, C. et al. Efficient gene transfer into human hepatocytes by baculovirus vectors. Proc. Natl. Acad. Sci. USA 92, 10099–10103 (1995).
Kost, T.A., Condreay, J.P., Ames, R.S., Rees, S. & Romanos, M.A. Implementation of BacMam virus gene delivery technology in a drug discovery setting. Drug Discov. Today 12, 396–403 (2007).
Ames, R. et al. BacMam recombinant baculoviruses in G protein–coupled receptor drug discovery. Receptors Channels 10, 99–107 (2004).
Boudjelal, M. et al. The application of BacMam technology in nuclear receptor drug discovery. Biotechnol. Annu. Rev. 11, 101–125 (2005).
Pfohl, J.L. et al. Titration of KATP channel expression in mammalian cells utilizing recombinant baculovirus transduction. Receptors Channels 8, 99–111 (2002).
Fonfria, E. et al. Cloning and pharmacological characterization of the guinea pig P2X7 receptor orthologue. Br. J. Pharmacol. 153, 544–556 (2008).
Shukla, S., Schwartz, C., Kapoor, K., Kouanda, A. & Ambudkar, S.V. Use of baculovirus BacMam vectors for expression of ABC drug transporters in mammalian cells. Drug Metab. Dispos. 40, 304–312 (2012).
Scott, M.J. et al. Efficient expression of secreted proteases via recombinant BacMam virus. Protein Expr. Purif. 52, 104–116 (2007).
Dukkipati, A., Park, H.H., Waghray, D., Fischer, S. & Garcia, K.C. BacMam system for high-level expression of recombinant soluble and membrane glycoproteins for structural studies. Protein Expr. Purif. 62, 160–170 (2008).
Ely, L.K., Fischer, S. & Garcia, K.C. Structural basis of receptor sharing by interleukin 17 cytokines. Nat. Immunol. 10, 1245–1251 (2009).
Liu, H. et al. The mechanism of shared but distinct CSF-1R signaling by the non-homologous cytokines IL-34 and CSF-1. Biochim. Biophys. Acta 1824, 938–945 (2012).
Lupardus, P.J. et al. Structural snapshots of full-length Jak1, a transmembrane gp130/IL-6/IL-6Rα cytokine receptor complex, and the receptor-Jak1 holocomplex. Structure 19, 45–55 (2011).
Deupi, X. et al. Stabilized G protein binding site in the structure of constitutively active metarhodopsin-II. Proc. Natl. Acad. Sci. USA 109, 119–124 (2012).
Baconguis, I., Bohlen, C.J., Goehring, A., Julius, D. & Gouaux, E. X-ray structure of acid-sensing ion channel 1–snake toxin complex reveals open state of a Na+-selective channel. Cell 156, 717–729 (2014).
Baconguis, I. & Gouaux, E. Structural plasticity and dynamic selectivity of acid-sensing ion channel–spider toxin complexes. Nature 489, 400–405 (2012).
Penmatsa, A., Wang, K.H. & Gouaux, E. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature 503, 85–90 (2013).
Chen, P.H., Chen, X., Lin, Z., Fang, D. & He, X. The structural basis of R-spondin recognition by LGR5 and RNF43. Genes Dev. 27, 1345–1350 (2013).
Liao, M., Cao, E., Julius, D. & Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 504, 107–112 (2013).
Lee, C.H. et al. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 511, 191–197 (2014).
Kawate, T. & Gouaux, E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681 (2006).
Hattori, M., Hibbs, R.E. & Gouaux, E. A fluorescence-detection size-exclusion chromatography-based thermostability assay for membrane protein precrystallization screening. Structure 20, 1293–1299 (2012).
Coric, T., Zheng, D., Gerstein, M. & Canessa, C.M. Proton sensitivity of ASIC1 appeared with the rise of fishes by changes of residues in the region that follows TM1 in the ectodomain of the channel. J. Physiol. 568, 725–735 (2005).
Cully, D.F. et al. Cloning of an avermectin-sensitive glutamate-gated chloride channel from Caenorhabditis elegans. Nature 371, 707–711 (1994).
Hibbs, R.E. & Gouaux, E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature 474, 54–60 (2011).
Berger, I., Fitzgerald, D.J. & Richmond, T.J. Baculovirus expression system for heterologous multiprotein complexes. Nat. Biotechnol. 22, 1583–1587 (2004).
Reeves, P.J., Callewaert, N., Contreras, R. & Khorana, H.G. Structure and function in rhodopsin: high-level expression of rhodopsin with restricted and homogeneous N-glycosylation by a tetracycline-inducible N-acetylglucosaminyltransferase I-negative HEK293S stable mammalian cell line. Proc. Natl. Acad. Sci. USA 99, 13419–13424 (2002).
Chae, P.S. et al. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat. Methods 7, 1003–1008 (2010).
Cooper, D.R. et al. Protein crystallization by surface entropy reduction: optimization of the SER strategy. Acta Crystallogr. D Biol. Crystallogr. 63, 636–645 (2007).
Magnani, F., Shibata, Y., Serrano-Vega, M.J. & Tate, C.G. Co-evolving stability and conformational homogeneity of the human adenosine A2a receptor. Proc. Natl. Acad. Sci. USA 105, 10744–10749 (2008).
Serrano-Vega, M.J., Magnani, F., Shibata, Y. & Tate, C.G. Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. USA 105, 877–882 (2008).
Hopkins, R. & Esposito, D. A rapid method for titrating baculovirus stocks using the Sf-9 Easy Titer cell line. Biotechniques 47, 785–788 (2009).
Ferris, M.M. et al. Evaluation of the Virus Counter for rapid baculovirus quantitation. J. Virol. Methods 171, 111–116 (2011).
Shen, C.F., Meghrous, J. & Kamen, A. Quantitation of baculovirus particles by flow cytometry. J. Virol. Methods 105, 321–330 (2002).
Dulbecco, R. & Vogt, M. Some problems of animal virology as studied by the plaque technique. Cold Spring Harb. Symp. Quant. Biol. 18, 273–279 (1953).
Al-Fageeh, M.B., Marchant, R.J., Carden, M.J. & Smales, C.M. The cold-shock response in cultured mammalian cells: harnessing the response for the improvement of recombinant protein production. Biotechnol. Bioeng. 93, 829–835 (2006).
Ho, Y.C., Chen, H.C., Wang, K.C. & Hu, Y.C. Highly efficient baculovirus-mediated gene transfer into rat chondrocytes. Biotechnol. Bioeng. 88, 643–651 (2004).
Hsu, C.S., Ho, Y.C., Wang, K.C. & Hu, Y.C. Investigation of optimal transduction conditions for baculovirus-mediated gene delivery into mammalian cells. Biotechnol. Bioeng. 88, 42–51 (2004).
Kumar, N., Gammell, P. & Clynes, M. Proliferation control strategies to improve productivity and survival during CHO-based production culture: a summary of recent methods employed and the effects of proliferation control in product secreting CHO cell lines. Cytotechnology 53, 33–46 (2007).
Pan, Y. et al. Efficient gene delivery into mammalian cells by recombinant baculovirus containing a hybrid cytomegalovirus promoter/Semliki Forest virus replicon. J. Gene Med. 11, 1030–1038 (2009).
Ramos, L. et al. Rapid expression of recombinant proteins in modified CHO cells using the baculovirus system. Cytotechnology 38, 37–41 (2002).
Sumitomo, Y. et al. Identification of a novel enhancer that binds Sp1 and contributes to induction of cold-inducible RNA-binding protein (cirp) expression in mammalian cells. BMC Biotechnol. 12, 72 (2012).
Wulhfard, S. et al. Mild hypothermia improves transient gene expression yields several fold in Chinese hamster ovary cells. Biotechnol. Prog. 24, 458–465 (2008).
Li, P., Slimko, E.M. & Lester, H.A. Selective elimination of glutamate activation and introduction of fluorescent proteins into a Caenorhabditis elegans chloride channel. FEBS Lett. 528, 77–82 (2002).
Backliwal, G. et al. Valproic acid: a viable alternative to sodium butyrate for enhancing protein expression in mammalian cell cultures. Biotechnol. Bioeng. 101, 182–189 (2008).
Barsoum, J., Brown, R., McKee, M. & Boyce, F.M. Efficient transduction of mammalian cells by a recombinant baculovirus having the vesicular stomatitis virus G glycoprotein. Hum. Gene Ther. 8, 2011–2018 (1997).
Andrell, J. & Tate, C.G. Overexpression of membrane proteins in mammalian cells for structural studies. Mol. Membr. Biol. 30, 52–63 (2013).
Aricescu, A.R., Lu, W. & Jones, E.Y. A time- and cost-efficient system for high-level protein production in mammalian cells. Acta Crystallogr. D Biol. Crystallogr. 62, 1243–1250 (2006).
Lee, J.E., Fusco, M.L. & Saphire, E.O. An efficient platform for screening expression and crystallization of glycoproteins produced in human cells. Nat. Protoc. 4, 592–604 (2009).
Wurm, F. & Bernard, A. Large-scale transient expression in mammalian cells for recombinant protein production. Curr. Opin. Biotechnol. 10, 156–159 (1999).
Chaudhary, S., Pak, J.E., Gruswitz, F., Sharma, V. & Stroud, R.M. Overexpressing human membrane proteins in stably transfected and clonal human embryonic kidney 293S cells. Nat. Protoc. 7, 453–466 (2012).
Reeves, P.J., Thurmond, R.L. & Khorana, H.G. Structure and function in rhodopsin: high level expression of a synthetic bovine opsin gene and its mutants in stable mammalian cell lines. Proc. Natl. Acad. Sci. USA 93, 11487–11492 (1996).
Reeves, P.J., Kim, J.M. & Khorana, H.G. Structure and function in rhodopsin: a tetracycline-inducible system in stable mammalian cell lines for high-level expression of opsin mutants. Proc. Natl. Acad. Sci. USA 99, 13413–13418 (2002).
Cockrell, A.S. & Kafri, T. Gene delivery by lentivirus vectors. Mol. Biotechnol. 36, 184–204 (2007).
Russell, W.C. Update on adenovirus and its vectors. J. Gen. Virol. 81, 2573–2604 (2000).
Dubensky, T.W. Jr. et al. Sindbis virus DNA-based expression vectors: utility for in vitro and in vivo gene transfer. J. Virol. 70, 508–519 (1996).
Nivitchanyong, T., Tsai, Y.C., Betenbaugh, M.J. & Oyler, G.A. An improved in vitro and in vivo Sindbis virus expression system through host and virus engineering. Virus Res. 141, 1–12 (2009).
Bennink, J.R. & Yewdell, J.W. Recombinant vaccinia viruses as vectors for studying T lymphocyte specificity and function. Curr. Top. Microbiol. Immunol. 163, 153–184 (1990).
Berglund, P. et al. Semliki Forest virus expression system: production of conditionally infectious recombinant particles. Biotechnology (NY) 11, 916–920 (1993).
Liljestrom, P. & Garoff, H. A new generation of animal cell expression vectors based on the Semliki Forest virus replicon. Biotechnology (NY) 9, 1356–1361 (1991).
Gonzales, E.B., Kawate, T. & Gouaux, E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature 460, 599–604 (2009).
Hattori, M. & Gouaux, E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 485, 207–212 (2012).
Jasti, J., Furukawa, H., Gonzales, E.B. & Gouaux, E. Structure of acid-sensing ion channel 1 at 1.9 Å resolution and low pH. Nature 449, 316–323 (2007).
Sobolevsky, A.I., Rosconi, M.P. & Gouaux, E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature 462, 745–756 (2009).
Hill, D.R., Aumiller, J.J., Shi, X. & Jarvis, D.L. Isolation and analysis of a baculovirus vector that supports recombinant glycoprotein sialylation by SfSWT-1 cells cultured in serum-free medium. Biotechnol. Bioeng. 95, 37–47 (2006).
Jarvis, D.L. Developing baculovirus-insect cell expression systems for humanized recombinant glycoprotein production. Virology 310, 1–7 (2003).
Wasilko, D.J. et al. The titerless infected-cells preservation and scale-up (TIPS) method for large-scale production of NO-sensitive human soluble guanylate cyclase (sGC) from insect cells infected with recombinant baculovirus. Protein. Expr. Purif. 65, 122–132 (2009).
Acknowledgements
We thank members of the Gouaux laboratory for helpful discussions. We are grateful to D. Goodman and G. Westbrook for encouragement and L. Vaskalis for assistance with figures. This work was supported by an Oregon Health and Science University Brain Institute Graduate Student Fellowship for Research on the Neurobiology of Disease (C.-H.L.), by a F32 Postdoctoral National Research Service Award (NRSA) from the US National Institute of Mental Health (NIMH) (K.H.W.), by a postdoctoral fellowship (Forschungsstipendium AL 1725-1/1) from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) (T.A.), by a F32 Postdoctoral NRSA from the US National Institute of General Medical Sciences (NIGMS) (D.P.C.), by the US National Institutes of Health (NIH) (E.G.) and by the Vollum Institute. E.G. is an investigator with the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Contributions
A.G. screened and optimized expression conditions for cASIC1 and GluCl. A.G., C.-H.L., K.H.W., J.C.M. and D.P.C. optimized the cell growth and virus amplification conditions. C.-H.L. designed the BacMam construct and performed initial characterization of the BacMam construct. I.B. and T.A. cloned and optimized the cASIC1 and GluCl pEG BacMam constructs, respectively. K.C.G. and S.F. provided the pVLAD construct, incubator configuration and consultations to optimize cell growth during the early stages of the project. All authors wrote and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Goehring, A., Lee, CH., Wang, K. et al. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protoc 9, 2574–2585 (2014). https://doi.org/10.1038/nprot.2014.173
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2014.173
This article is cited by
-
A nanobody-based strategy for rapid and scalable purification of human protein complexes
Nature Protocols (2024)
-
Structural basis for excitatory neuropeptide signaling
Nature Structural & Molecular Biology (2024)
-
Transport and inhibition mechanisms of human VMAT2
Nature (2024)
-
Oligomeric organization of membrane proteins from native membranes at nanoscale spatial and single-molecule resolution
Nature Nanotechnology (2024)
-
Cryo-EM structures of pannexin 1 and 3 reveal differences among pannexin isoforms
Nature Communications (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
