
Abstract
Human brain development exhibits several unique aspects, such as increased complexity and expansion of neuronal output, that have proven difficult to study in model organisms. As a result, in vitro approaches to model human brain development and disease are an intense area of research. Here we describe a recently established protocol for generating 3D brain tissue, so-called cerebral organoids, which closely mimics the endogenous developmental program. This method can easily be implemented in a standard tissue culture room and can give rise to developing cerebral cortex, ventral telencephalon, choroid plexus and retinal identities, among others, within 1–2 months. This straightforward protocol can be applied to developmental studies, as well as to the study of a variety of human brain diseases. Furthermore, as organoids can be maintained for more than 1 year in long-term culture, they also have the potential to model later events such as neuronal maturation and survival.
Similar content being viewed by others


Introduction
In vitro methods to model human development and disease are part of a rapidly expanding field of stem cell biology with major therapeutic implications1. Organoid protocols stand at the forefront of these technologies, as these 3D approaches more accurately reproduce in vivo developmental events leading to more precise in vitro models2,3. Organoids have already been developed for several organ systems, including retina4, intestine5, thyroid6, liver7, pituitary8, inner ear9, kidney10,11,12 and brain13. However, only a handful of methods exist for generating such tissues from human pluripotent stem cells (hPSCs)13,14,15,16,17. In this protocol, we describe the methodology that was recently published for generating brain organoid tissues from hPSCs13.
Development of the protocol
The method described here builds on an extensive foundation of protocols for neural differentiation, 3D tissue culture and tissue engineering. Cerebral organoids develop through intrinsic self-organizing processes upon timely application of components and culture environments that had previously been described individually. Thus, the method is an amalgamation of previous methods, combined in a specific manner to address two main objectives: (i) the establishment of neural identity and differentiation and (ii) the recapitulation of 3D structural organization.
Establishment of brain identity
The first goal of the protocol is induction and differentiation of neural tissue. This involves identification of media formulations and additives to drive neural identity and to stimulate brain development in vitro. To achieve this, a number of medium formulations were tested at various time points. Rather than describing the multitude of tested combinations, we focus here on the successful outcome.
Neural tissue develops in vivo from a germ layer called the ectoderm18. Similarly, PSCs in vitro can be stimulated to develop germ layers, including ectoderm, within aggregates called embryoid bodies (EBs)19. A number of previous studies have described successful differentiation of EBs in embryonic stem cell (ESC) medium with decreased basic fibroblast growth factor (bFGF)20 and high-dose rho-associated protein kinase (ROCK) inhibitor to limit cell death21. Similarly, cerebral organoids develop from EBs grown initially in ESC medium with low bFGF and ROCK inhibitor.
Subsequent neural induction of EBs follows a minimal medium formulation very similar to that established by Zhang et al.22,23 for the induction of neural rosettes, a 2D polarized organization of neuroepithelial cells. However, for the generation of cerebral organoids, EBs are kept in suspension, leading to uniform neural ectoderm formation along the outer surface of EBs, whereas inner non-neural mesendodermal tissues do not develop.
Neural ectoderm in vivo establishes radially organized neuroepithelia that expand to form various brain structures. Similarly, organoids placed in a differentiation medium that supports both neural progenitors and their progeny display this developmental progression in vitro. Medium formulation at this stage is influenced by a large body of neural stem cell (NSC) protocols24,25 and neural rosette methods26,27. These methods have demonstrated the importance of Neurobasal medium and B27 supplement for neuronal differentiation and survival28. Empirical testing of medium formulations from various rosette protocols led to the inclusion of additional supplements such as 2-mercaptoethanol27 and insulin29, which seem to have a positive effect in maintaining NSCs.
Finally, retinoic acid has previously been described to be secreted from the brain meninges and promote neural differentiation30. However, retinoic acid is a potent caudalizing factor in vivo31. Therefore, as we sought to limit the application of exogenous patterning factors, this component is not included at early stages and it is only included in differentiation medium at later stages in the form of vitamin A provided in the B27 supplement.
Establishment of 3D spatial organization
The second objective of the method was achieving a 3D spatial organization that could recapitulate the development of various brain regions. A number of recent studies have demonstrated the enormous self-organizing capacity of tissues developed from PSCs, including certain neural tissues32. Therefore, we focused on providing a permissive environment for 3D self-organization.
Neural ectoderm in vitro can spontaneously acquire a radial organization reminiscent of neuroepithelium, as in the case of neural rosettes. Similarly, neural ectoderm of EBs spontaneously establishes apicobasal polarity to form the neuroepithelium. However, in the absence of the basement membrane normally present in vivo, this epithelium lacks proper orientation and fails to form a continuous epithelium33. We therefore tested the effect of providing a structural support to promote continuity and proper orientation.
Various studies have demonstrated the formation of complex organized epithelia within hydrogels composed of extracellular matrix proteins34. In particular, intestinal stem cells embedded in Matrigel have recently been shown to generate large intestinal epithelial 3D tissues, termed intestinal organoids5. Similarly, we tested the application of this approach to neural ectoderm-induced EBs. Shortly after embedding, large buds of continuous neuroepithelium protrude from the larger EBs and contain fluid-filled cavities reminiscent of brain ventricles indicative of proper apicobasal orientation.
The final key element of the protocol is the application of agitation. It became clear that, although Matrigel promoted neuroepithelial bud expansion, organoids quickly grew beyond the limits of stationary diffusion of oxygen and nutrients, as evidenced by dark necrotic tissue at the center of organoids. As agitating bioreactors have proven useful in a number of tissue engineering applications2,3,35, we tested the effect of a spinning bioreactor to better promote diffusion. This markedly improved tissue survival and further development. Other types of agitation are also acceptable, and the method described here also details the use of an orbital shaker as an alternative to the spinning bioreactor.
Comparison with alternative methods
A number of methods exist for growth and differentiation of NSCs, the vast majority of which are performed in monolayer culture. For decades, NSC lines have been used to generate particular neural and non-neural cell types for potential therapeutic applications36,37. However, these lines lack important hallmarks of stem cells of the developing brain, such as apicobasal orientation.
More recently, neural rosettes have been derived from PSCs that properly recapitulate this apicobasal organization, thus forming a radially organized pattern in 2D, much like the neural tube epithelium13,14,15,16,17,22,26. Neural rosettes recapitulate many aspects of brain development, including proper lineage progression13,38 and timed neuronal specification18,27; however, they lack the complex organization generated using a 3D approach such as the cerebral organoid method. Organoids are more heterogeneous than rosettes in 2D, but 3D neuroepithelia are more continuous, they expand to form defined progenitor zones and they develop several brain region identities; thus, they better recapitulate the complex interplay of different regions and structures.
Methods of generating neural tissue in 3D other than the method described here are quite limited. Neurospheres are 3D aggregates of neural progenitors often used as an assay to evaluate proliferative capacity of neural progenitors19,37. However, they lack clear organization and therefore suffer from many of the limitations of NSC lines grown in 2D. The most similar approach to the method described here is the SFEBq (serum-free floating culture of embryoid body–like aggregates with quick reaggregation) method 20,21. This method has been successfully applied to the derivation of a number of individual brain regions, including the retina4,21, cerebral cortex21,22,23 and pituitary8,24,25.
There are certain similarities between the SFEBq and cerebral organoid methods, particularly the structure of dorsal telencephalic tissues generated21,26,27, but the medium formulations differ considerably and the developmental progression and therefore timing is quite distinct. Most notably, the SFEBq method makes use of growth factors to stimulate neural differentiation and subsequent differentiation into specific regions. Cerebral organoids instead spontaneously acquire various brain tissue identities and therefore establish multiple regions within a single organoid.
The SFEBq method has been described to exhibit limited continuity and expansion of neuroepithelial tissue21,28,33. Instead, the method described here generates brain organoids with large continuous neuroepithelium that arises when embedded in Matrigel, which, along with agitation, distinguishes the organoid method. This issue has recently been resolved in the SFEBq method, and interestingly the solution seems to be an addition of extracellular matrix proteins, in the form of low-concentration dissolved Matrigel27,39 or laminin/entactin proteins29,33. Other modifications have also recently been published, including growth in a high-oxygen environment and the addition of serum, lipids and heparin30,39, the latter of which is also an important component of the organoid method.
Applications and limitations of the protocol
The following protocol can be used for a variety of developmental and disease studies. It is particularly suited to examining biological questions that would benefit from a human model system. For example, we have used cerebral organoids to examine cell division orientation in human radial glial stem cells13,31, a process thought to be uniquely regulated in humans compared with mice32,40. Similarly, brain organoids can be used to examine fate determination of NSCs or their intermediate progeny. More generally, the organoids can be used to examine tissue morphogenesis, early embryonic ectodermal fate determination and neuroepithelial polarity establishment.
Cerebral organoids represent a novel system to interrogate the mechanisms of human neurological conditions that have been difficult or impossible to examine in mice and other model organisms. We have used brain organoids to examine the cell biological basis of a form of microcephaly, a disorder involving small brain size13,33,41. Similarly, a variety of neurological disorders could be examined in cerebral organoids. Broadly speaking, this system is perhaps most suited to examining neurodevelopmental disorders, as it best recapitulates the early developing brain (first trimester, on the basis of histological comparisons). These include disorders with clear morphological abnormalities but also more common disorders such as autism, intellectual disability and epilepsy34,42.
Finally, it is important to keep in mind the limitations of using such a system to examine brain development and disease. As in all in vitro systems, the method lacks surrounding embryonic tissues that are important for the interplay of neural and non-neural tissue cross-talk. Specifically, the lack of the overlying meninges and the vasculature that it provides severely limits the growth potential of the organoids. This contributes to certain stochastic growth patterns depending on the availability of nutrients and the lack of body axes to pattern the neural tissue. Therefore, organoids show marked variability, particularly between preparations. Thus, to consistently detect phenotypes, for example, in the case of genetic mutations, defects must be robust enough to lie outside the normal range. In addition, proper controls for this variability must be included, such as control organoids prepared at the same time and grown in the same medium, and, if possible, comparison of cellular phenotypes within single organoids.
Experimental design
Although we have previously generated cerebral organoids from both mouse and hPSCs13, the method described here is specific for hPSCs. We have successfully tested both ESCs and induced PSCs (iPSCs), as well as feeder-dependent and feeder-independent cell lines. PSCs are first dissociated to single cells and allowed to reaggregate to form EBs (Fig. 1). Previous studies have demonstrated the successful generation of EBs in 96-well U-bottom plates coated with nonadhesive compounds43. Such coated plates are commercially available and can be used to reliably generate EBs of homogeneous size and morphology.

The protocol begins with the generation of EBs from human PSCs in a 96-well U-bottom plate. The day on which EBs are made is day 0. Generation of EBs is outlined in Step 1. Feeding and monitoring the EBs is described in Steps 2–4. Typically, on day 6, the EBs are transferred to a 24-well plate containing neural induction medium, as described in Step 5. Feeding and monitoring of neural induction is described in Steps 6 and 7. On day 11, neuroectodermal tissues are then transferred to droplets of Matrigel on a sheet of dimpled Parafilm, as described in Steps 8–17, and then grown in a 60-mm dish. Monitoring of these tissues is described in Steps 18 and 19. Finally, Matrigel droplets are transferred to the spinning bioreactor on day 15, as described in Step 20, and further maintained as described in Step 21.
EBs are then subjected to neural induction in a minimal medium that does not support the growth of endoderm and mesoderm, and instead allows only the neuroectoderm to develop. The neuroectodermal tissues are then transferred to a floating droplet of Matrigel, which promotes outgrowth of neuroepithelial buds that expand and contain fluid-filled lumens. Finally, the tissues are transferred to a spinning bioreactor, or alternatively to an orbital shaking plate, which promotes improved nutrient and oxygen exchange to allow more extensive growth and further development into defined brain regions.
Materials
REAGENTS
Cells
-
hPSCs or iPSCs. We have successfully used H9 human ESCs (hESCs), feeder-dependent or feeder-independent (Wisconsin International Stem Cell (WISC) Bank, Wicell Research Institute, WA09 cells) and reprogrammed feeder-dependent iPSCs (Systems Biosciences, cat. no. SC101A-1)
Caution
The use of human tissues and human stem cells must adhere to institutional and funding body regulations, as well as to relevant ethical guidelines.
-
Growth-arrested irradiated mouse embryonic fibroblast feeder cells, if using feeder-dependent stem cells (mouse embryonic fibroblasts (MEFs); GlobalStem, cat. no. GSC-6001G)
Growth media and supplements
-
mTeSR1 medium (Stem Cell Technologies, cat. no. 05850)
-
DMEM-F12 (Invitrogen, cat. no. 11330-032 or 31330-038, depending on location)
-
Knockout serum replacement (KOSR; Invitrogen, cat. no. 10828-028)
-
hESC-quality FBS (it should be tested for compatibility with hESCs; Gibco, cat. no. 10270-106)
-
GlutaMAX (Invitrogen, cat. no. 35050-038)
-
MEM non-essential amino acids (MEM-NEAA; Sigma, cat. no. M7145)
-
2-Mercaptoethanol (Merck, cat. no. 8057400005)
-
bFGF (FGF2; Peprotech, cat. no. 100-18B)
Critical
We have not tested bFGF from other vendors for this protocol.
-
BSA (AppliChem, cat. no. 9048-46-8)
-
Sterile water
-
Heparin (Sigma, cat. no. H3149)
-
ROCK inhibitor Y27632 (Millipore, cat. no. SCM075)
-
N2 supplement (Invitrogen, cat. no. 17502048)
-
B27 – vitamin A supplement (Invitrogen, cat. no. 12587010)
-
B27 + vitamin A supplement (Invitrogen, cat. no. 17504044)
-
Penicillin-streptomycin (Sigma, cat. no. P0781)
-
Neurobasal medium (Invitrogen, cat. no. 21103049)
-
Insulin solution (Sigma, cat. no. I9278-5ML)
Enzymes and other reagents
-
Growth factor–reduced Matrigel (BD Biosciences, cat. no. 356230)
-
Gelatin (Sigma, cat. no. G1890-100G)
-
Collagenase IV (Invitrogen, cat. no. 17104-019)
-
Sterile PBS (Invitrogen, cat. no. 14040-091)
-
EDTA (Sigma-Aldrich, cat. no. E6758)
-
Sterile D-PBS without calcium and magnesium (Invitrogen, cat. no. 14190-094)
-
Matrigel (BD Biosciences, cat. no. 356234)
-
Dispase (Invitrogen, cat. no. 17105-0412q)
-
Spray bottle containing 70% (vol/vol) ethanol
-
Trypsin/EDTA solution, 0.05% (wt/vol) (Gibco, cat. no. 25300-054)
-
Trypsin inhibitor (Sigma-Aldrich, cat. no. T6414-100ML)
-
Accutase (Sigma-Aldrich, cat. no. A6964)
-
Trypan blue (Bio-Rad, cat. no. 145-0021)
-
Paraformaldehyde (PFA; Sigma-Aldrich, cat. no. 158127)
-
Sucrose (Sigma-Aldrich, cat. no. S7903)
EQUIPMENT
-
CO2 incubators (Thermo Scientific, cat. no. 51026280)
-
Biological safety cabinet (Thermo Scientific, cat. no. 51022482)
-
Six-well tissue culture dishes (Corning, cat. no. 353046)
-
Cell lifter (Corning, cat. no. 3008)
-
Rainin Pipet Plus (P1000, P200 and P10)
-
Sterile filter pipette tips (1 ml, 200 and 10 μl; Sigma-Aldrich, cat. nos. A3348, A3098 and A2473, respectively)
-
Sterile microcentrifuge tubes (1.5-ml size; Fisher Scientific, cat. no. 05-408-129)
-
Sterile 10-ml syringe without needle (Sigma, cat. no. Z248029)
-
Syringe filter, 0.2 μm (Sigma-Aldrich, cat. no. Z259969)
-
Stericup 0.2-μm filter unit (500 and 250 ml; Millipore, cat. nos. SCGVU02RE SCGVU05RE, respectively)
-
U-bottom ultralow attachment plates, 96 well (Corning, cat. no. CLS7007)
-
Conical tubes, 15 ml (Fisher Scientific, cat. no. 05-527-90)
-
Ultralow attachment plates, 24 well (Corning, cat. no. CLS3473)
-
Parafilm (Sigma-Aldrich, cat. no. P7793)
-
Tissue culture dish, 60 mm (Sigma-Aldrich, cat. no. CLS430589)
-
Spinner flask, 125 ml (Corning, cat. no. 4500-125)
-
Stir plate (2mag, bioMIXdrive and bioMIXcontrol)
-
Orbital shaker (IKA, cat. no. 0009019200)
-
Pipetboy (Integra Biosciences, cat. no. 155 000)
-
Serological pipettes, 10 ml (BD Falcon, cat. no. 357551)
-
Sterilized scissors
-
Water bath, 37 °C (Fisher Scientific, Isotemp water bath)
-
Stereomicroscope (Zeiss, Stemi 2000)
-
Inverted contrasting tissue culture microscope (Leica, DMIL)
-
Automated cell counter (Bio-Rad, TC10)
-
Benchtop centrifuge (Thermo Scientific, Heraeus Multifuge 1s)
-
Sterile standard forceps (Fine Science Tools, cat. no. 11000)
-
Standard 24-well tissue culture plate (Sigma-Aldrich, cat. no. CLS3527)
-
Weighing dishes (Sigma-Aldrich, cat. no. Z154873)
-
Scalpel blade (Sigma-Aldrich, cat. no. S2771)
-
Isopentane (2-methylbutane; Sigma-Aldrich, cat. no. M32631)
-
Dry ice
-
Low-temperature thermometer (Sigma-Aldrich, cat. no. Z257400)
-
Laboratory spatula (Sigma-Aldrich, cat. no. S3897)
-
Cryostat (Leica)
REAGENT SETUP
Feeder-dependent hPSC lines
-
Culture hPSCs using standard procedures in a 5% CO2 incubator at 37 °C. Briefly, maintain feeder-dependent hESCs or hiPSCs on growth-arrested MEFs according to a protocol modified rom WISC Bank protocols (http://www.wicell.org/home/support/stem-cell-protocols/stem-cell-protocols.cmsx). Plate g-irradiated MEFs on gelatin-coated (0.1% (wt/vol) gelatin) six-well BD Falcon plates at a density of 1.87 × 105 cells per well the day before splitting or thawing hESCs or hiPSCs. Passage feeder-dependent PSCs using 0.1% (wt/vol) collagenase IV in DMEM-F12 medium for 5–10 min, followed by scraping with a cell lifter to remove intact colonies and triturate with a 1-ml pipette tip to obtain smaller colonies before plating. Maintain feeder-dependent PSCs in hESC medium with a 20 ng ml−1 final concentration of bFGF.
Feeder-free hESCs
-
Maintain feeder-free hESCs in mTeSR1 medium and culture them on Matrigel-coated plates according to WISC Bank protocols. Briefly, dissolve low-growth-factor Matrigel in DMEM-F12 and use a volume containing 0.5 mg of Matrigel to coat an entire six-well BD Falcon plate. Passage feeder-independent hESCs using 0.5 mM EDTA in sterile D-PBS without calcium and magnesium.
Reconstitution and storage of growth factors and other additives
-
Prepare a 10% (wt/vol) solution of BSA by dissolving 1 g in 10 ml of sterile water. This can be stored at −20 °C for 1–2 years. Reconstitute 50 μg of bFGF in 5 ml of sterile PBS + 0.1% (wt/vol) final concentration of BSA to obtain a 10 μg ml−1 solution. Make 25-μl aliquots and store them at −20 °C for up to 6 months. Avoid repeated freezing and thawing. Reconstitute heparin in sterile PBS to a final stock concentration of 1 mg ml−1 and store it at 2–8 °C for up to 2 years. Reconstitute 5 mg of ROCK inhibitor in 2.96 ml of sterile water to obtain a final concentration of 5 mM. Make 150-μl aliquots and store them at −20 °C for up to 1 year. Aliquot and store N2 and B27 supplements at −20 °C for up to 1 year.
Aliquotting Matrigel for Matrigel droplets
-
Thaw the Matrigel on ice at 4 °C overnight. Precool 1-ml pipette tips and ten microcentrifuge tubes at −20 °C for 10–15 min. By using cold pipette tips, pipette the Matrigel up and down on ice and in a sterile hood before transferring 500 μl to each tube. Store the aliquots at −20 °C for up to 1 year. Avoid repeated freezing and thawing.
Critical
Matrigel will solidify at room temperature (22–25 °C), so it is important that all materials coming in contact with the solution be kept cold and that aliquotting be done quickly to minimize time at room temperature.
Dispase solution
-
Dissolve 5 mg of dispase in 5 ml of DMEM-F12, and filter it using a syringe and a 0.2-μm syringe filter. This solution is stable at 2–8 °C for up to 2 weeks, or larger quantities can be made and aliquots can be stored at −20 °C for up to 4 months.
hESC medium
-
For ∼500 ml of medium, combine 400 ml of DMEM-F12, 100 ml of KOSR, 15 ml of ESC-quality FBS, 5 ml of GlutaMAX, 5 ml of MEM-NEAA and 3.5 μl of 2-mercaptoethanol. Filter it using a vacuum-driven 0.2-μm Stericup filter unit. This can be stored for up to 2 weeks at 2–8 °C. Add bFGF to a final concentration of 20 ng ml−1 for standard hESC or hiPSC feeder-dependent culture, or for low-bFGF hESC medium, add bFGF to a final concentration of 4 ng ml−1.
Critical
Add bFGF immediately before use only to the volume needed.
Neural induction medium
-
Combine DMEM-F12 with 1% (vol/vol) N2 supplement, 1% (vol/vol) GlutaMAX supplement and 1% (vol/vol) MEM-NEAA. Add heparin (final concentration of 1 μg ml−1) and filter it using a vacuum-driven 0.2-μm filter unit. Store it at 2–8 °C for up to 2 weeks.
Cerebral organoid differentiation medium
-
For ∼250 ml of medium, combine 125 ml of DMEM-F12, 125 ml of Neurobasal medium, 1.25 ml of N2 supplement, 62.5 μl of insulin, 2.5 ml of GlutaMAX supplement, 1.25 ml of MEM-NEAA and 2.5 ml of penicillin-streptomycin. Prepare a 1:100 dilution of 2-mercaptoethanol in DMEM-F12 and add 87.5 μl of this to the medium. Add 2.5 ml of B27 supplement.
Critical
B27 should not contain vitamin A (retinoic acid) during the initial stages of growth in Matrigel. However, once droplets are transferred to the orbital shaker or spinner flask, B27 should include vitamin A. Filter the medium using a vacuum-driven 0.2-μm filter unit and store it at 2–8 °C for up to 2 weeks.
Reagents for organoid analysis and cryosectioning
-
Prepare a 4% (wt/vol) PFA solution by dissolving 4 g of PFA in 80 ml of PBS at 60 °C by adding a few drops of 1 N NaOH until the powder dissolves. Adjust the pH to 7.4 and bring the final volume up to 100 ml. Divide it into aliquots and store it at −20 °C. Prepare a 30% (wt/vol) sucrose solution by dissolving 30 g of sucrose in a 100-ml final volume of PBS at 37 °C. Prepare gelatin/sucrose embedding solution by first dissolving 100 g of sucrose in 1 liter of PBS at 37 °C to prepare a 10% (wt/vol) sucrose solution. Store the sucrose solutions at 4 °C. Dissolve 7.5 g of gelatin in 100 ml of 10% (wt/vol) sucrose solution at 37 °C to obtain a 7.5% gelatin/10% sucrose embedding solution (wt/vol). Divide the solution into aliquots and store it at −20 °C.
EQUIPMENT SETUP
Spinning bioreactor
-
Install a low-speed stir plate appropriate for use in a CO2 incubator by first spraying it with ethanol to sterilize and then placing it on the bottom shelf of a standard tissue culture incubator. Be sure that the shelves above are positioned with enough room to fit the spinner flask on the stir plate comfortably. Move the power cable or controller cable to the side so that it can exit the incubator without hanging in front of tissue culture dishes. Hold it in place with a piece of tape and check that the incubator doors can still close securely.
Orbital shaker
-
Spray a standard orbital shaker with ethanol and place it on a shelf in the tissue culture incubator, making sure that the shelf above is positioned with enough room for tissue culture dishes to be placed on the shaker. Move the power cable to the side so that it can exit the incubator without hanging in front of tissue culture dishes. Hold it in place with a piece of tape and check that the incubator doors can still close securely.
Procedure
Making EBs
Timing 1–2 h
-
1
Grow hESC or iPSC colonies in one well of a six-well plate until they are 70–80% confluent. All or part of the well can be used to make EBs. Typically, one well will yield approximately an entire 96-well plate of EBs. Follow option A to generate EBs from feeder-dependent PSCs and option B for EB generation from feeder-independent hESCs. We have not observed a difference in organoids generated from either feeder-dependent or feeder-independent PSCs.
-
A
From feeder-dependent PSCs
-
i
Wash the cells by removing the hESC medium and replacing it with 1 ml of D-PBS without calcium and magnesium. Remove D-PBS and add 1 ml of dispase solution to each well of a six-well plate, and then place the cells back in the incubator for 20–30 min. The colony edges should curl off the plate, being only attached at the center of the colony. This can take up to 40 min.
-
ii
Remove the dispase solution and add 1 ml of low-bFGF hESC medium. Tap the dish rigorously to remove the colonies from the dish without removing MEFs and differentiated cells. Transfer the medium containing intact colonies to a 15-ml conical tube, being careful to limit the disruption of colonies. Allow the colonies to settle to the bottom of the tube for ∼1 min.
-
iii
Gently aspirate the supernatant containing single cells and MEFs, being careful not to disturb the settled colonies. Add another 1 ml of low-bFGF hESC medium and again allow the colonies to settle; remove the supernatant.
-
iv
Resuspend the colonies in 1 ml of trypsin/EDTA and incubate the tube for 2 min at 37 °C. Add 1 ml of trypsin inhibitor and triturate the mixture using a 1-ml pipette tip until the solution becomes cloudy with single cells. Take two replicates of 5 μl for cell counting, and then add 8 ml of low-bFGF hESC medium.
-
v
Centrifuge the cells at 270g for 5 min at room temperature, and in the meantime count live cells by adding an equal volume of trypan blue to mark dead cells. Count the cells using a hemocytometer or an automated cell counter. Use the average of the two replicates to calculate subsequent steps.
-
vi
Resuspend the cells first in 1 ml of low-bFGF hESC medium with ROCK inhibitor (1:100, final concentration 50 μM). Pipette up and down to ensure a single-cell suspension. Then add an additional appropriate volume of low-bFGF hESC medium with ROCK inhibitor to obtain 9,000 live cells per 150 μl.
-
vii
Plate 150 μl in each well of a low-attachment 96-well U-bottom plate and place it back in the incubator.
-
i
-
B
From feeder-independent hESCs
-
i
Wash the cells with 1 ml of D-PBS without calcium and magnesium and add 600 μl of 0.5mM EDTA solution in D-PBS without calcium and magnesium for each well of the six-well plate. Place the cells back in the incubator for 4 min.
-
ii
Gently aspirate the EDTA solution without disturbing the colonies, and add 1 ml of Accutase. Place the cells back in the incubator for another 4 min.
-
iii
Use a 1-ml pipette tip to spray the colonies with 1 ml of mTeSR1 medium to detach them from the dish. Transfer 2 ml to a 15-ml conical tube and triturate the mixture using a 1-ml pipette tip until the solution becomes cloudy with single cells. Take two repetitions of 5 μl for cell counting, and then add another 3 ml of mTeSR1 medium and mix.
-
iv
Centrifuge the cells at 270g for 5 min at room temperature, and in the meantime count live cells by adding an equal volume of trypan blue to mark dead cells. Count the cells using a hemocytometer or an automated cell counter. Use the average of the two replicates to calculate subsequent steps.
-
v
Resuspend the cells first with 1 ml of low-bFGF hESC medium with ROCK inhibitor (1:100, final concentration 50 μM). Pipette up and down to ensure a single-cell suspension. Next, add an additional appropriate volume of low-bFGF hESC medium with ROCK inhibitor to obtain 9,000 live cells per 150 μl.
-
vi
Plate 150 μl in each well of a low-attachment 96-well U-bottom plate and place it back in the incubator.
-
i
Critical Step
The morphology of stem cell colonies is crucial to the success of cerebral tissue formation. The colonies should have no evidence of differentiation and should display optimal features of pluripotency (Fig. 2).
Figure 2: Progression of cerebral organoid development from human PSCs. 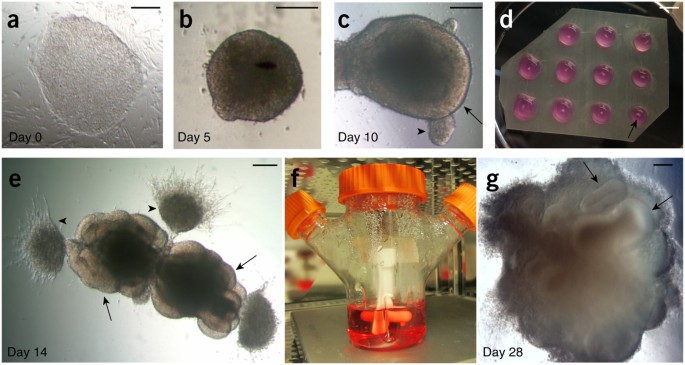
(a) A colony of feeder-dependent human ESCs showing typical pluripotent morphology with clear boundaries and a uniform texture. (b) An EB at day 5 showing evidence of ectodermal differentiation, as indicated by the presence of brightened surface tissue, whereas the center is quite dark with dense non-ectodermal tissue. The EB also has a smooth surface, indicating healthy tissue. (c) An early organoid at day 10 showing smooth edges and bright optically translucent surface tissue consistent with neuroectoderm (arrow). This organoid also contains small buds of ectodermal tissue that is not organized radially (arrowhead). (d) Image of the neuroectodermal tissues embedded in Matrigel droplets on a sheet of dimpled Parafilm. The tissues are visible as small white specks within the droplet (arrow). (e) An organoid at day 14, after embedding in Matrigel, showing evidence of neuroepithelial bud outgrowth (arrows) that are optically clear and in several cases surround a visible lumen. Other outgrowths and migrating cells are also visible (arrowheads) that are not neuroepithelial. (f) The spinning bioreactor setup in the tissue culture incubator. Organoids are visible within the bioreactor as small white floating specks. (g) An organoid at day 28 of the protocol, revealing many large neural tissues (arrows) that have greatly expanded once embedded in the Matrigel. Scale bars, 200 μm (a–c,e,g) and 5 mm (d).
-
A

Feeding EBs and initiation of germ layer differentiation
Timing 5–7 d
-
2
Observe the plate under the tissue culture microscope 24 h later. Small EBs with clear borders should be visible, although many dead cells will decorate the area around the EB. This is completely normal and will not disturb the formation of the EB at the center. Continue to culture EBs in the tissue culture incubator at 37 °C and 5% CO2.
-
3
Feed the EBs every other day by gently aspirating approximately half of the medium without disturbing the EB at the bottom of the well. Add an additional 150 μl of fresh medium (the final volume will be >150 μl, but the exact amount is not important). Include ROCK inhibitor (1:100) and low-bFGF medium (4 ng ml−1) until EBs begin to brighten or are >350–400 μm in diameter. Size measurements can be performed using an inverted microscope equipped with a camera and measurement software. Typically, ROCK inhibitor and low-bFGF medium are included only for the first 4 d.
-
4
While EBs are 350–600 μm in diameter, feed EBs every other day, as described in Step 3 using hESC medium, but do not include ROCK inhibitor or bFGF.
Induction of primitive neuroepithelia
Timing 4–5 d
-
5
When EBs are ∼500–600 μm in diameter and begin to brighten and have smooth edges (typically day 6; Figs. 2b and 3), transfer each EB with a cut 200-μl pipette tip to one well of a low-attachment 24-well plate containing 500 μl of neural induction medium (Fig. 1), being careful not to disrupt the EB. Continue culturing EBs.
Figure 3: Examples of suitable and suboptimal organoids at various stages. 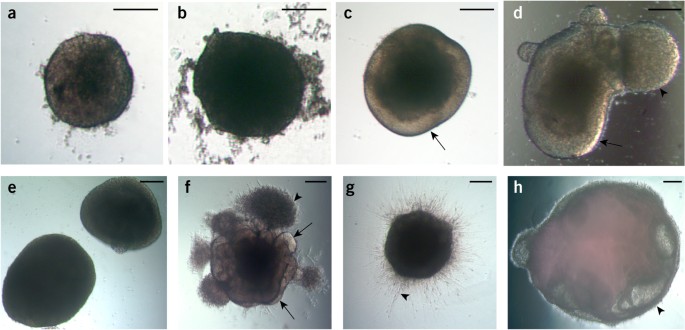
(a) An example of an optimal EB at day 5 showing brightening and clearing around the surface and with smooth edges. (b) An example of an unsuitable EB lacking optical clearing and with large amounts of cell debris, despite its large size. (c) An organoid in neural induction medium showing clear radially organized optically translucent neuroectoderm (arrow). (d) An example of an acceptable organoid also showing evidence of optically clear neuroectoderm (arrow) but also a large bud of translucent ectoderm that is not radially organized (arrowhead). This bud, although not ideal, will not affect development of the neighboring neuroectodermal tissue. (e) An example of failed neural induction. The EBs are too large and lack optically translucent, radially organized neuroectoderm. (f) An ideal organoid soon after Matrigel embedding, showing many buds of neuroepithelial (arrows) and non-neuroepithelial cells, which have migrated into the Matrigel (arrowhead). (g) An organoid that has failed to produce neuroepithelial buds, instead displaying extended cell processes (arrowhead) consistent with direct neural differentiation. (h) An example of a failed organoid after several weeks of differentiation showing large fluid-filled cysts that lack a thickened neuroepithelium (arrowhead). Scale bars, 200 μm.
Critical Step
Cut a 200-μl pipette tip with sterile scissors to obtain an opening of 1–1.5 mm in diameter. Be sure that the opening is not too small, as this will disrupt the EB, but also not too large, as this will make it difficult to suck the EB into the pipette tip. Do not attempt to scoop the EB out of the well with a spatula or other tools, as this will damage the EB.
-
6
Feed the EBs by adding another 500 μl of neural induction medium 48 h after transferring them to the 24-well plate.
-
7
Observe the EBs on the tissue culture microscope after a further 2 d. EBs should be brighter around the outside, indicating neuroectodermal differentiation. Once these regions begin to show radial organization of a pseudostratified epithelium consistent with neuroepithelium formation (Fig. 2c), which should happen after 4–5 d in neural induction medium, proceed to Step 8 to transfer the aggregates to Matrigel droplets (Fig. 1).
Critical Step
Healthy cell aggregates should have smooth edges. Neuroepithelium develops on the outer surface and is quite optically translucent (Figs. 2c and 3c,d). Occasionally, tissues may exhibit outgrowths or buds of optically translucent tissue that is not radially organized (Figs. 2c and 3d). Although it is not ideal, we have not noticed a long-term effect on organoid formation when these buds appear as long as radial organization is present in other regions of the tissue. Importantly, these nonradial regions can become radial if left in neural induction for 1–2 more days, but with the risk that other already radial regions may begin to shrink and lose the ability to form neuroepithelial buds if not transferred to Matrigel (Step 8) in a timely manner.
Critical Step
Be sure to move to Step 8 to transfer tissues to Matrigel when neuroepithelium appears. Do not transfer the tissues too late, as this can affect later morphology of cerebral tissues.

Transferring neuroepithelial tissues to Matrigel droplets
Timing 1–2 h
-
8
Thaw Matrigel on ice at 4 °C for 1–2 h. We find that a microcentrifuge tube containing 500 μl of Matrigel will thaw in this time frame if it is kept floating in a bath of ice and water.
-
9
Prepare dimpled Parafilm substrate for the generation of Matrigel droplets by layering a square of Parafilm over an empty tip tray for size 200-μl tips. Press your gloved finger into the Parafilm over each hole in the tip tray to create small dimples in the Parafilm.
Caution
Parafilm cannot be properly sterilized, as it cannot be autoclaved. Therefore, make sure to use Parafilm kept in a clean environment, and spray your gloves and the Parafilm with 70% (vol/vol) ethanol before preparing dimples. Antibiotics are also included in the medium at this stage to prevent contamination.
-
10
Make a grid of 4 × 4 dimples (16 total) and trim the Parafilm with sterile scissors to a small square containing this grid. Place the square of Parafilm into a 60-mm tissue culture dish.
Critical Step
A grid of 4 × 4, but not larger, will fit in the 60-mm dish. Therefore, a total of 16 droplets are placed in each 60-mm dish.
-
11
Use a cut 200-μl tip to transfer the neuroepithelial tissues one by one to each dimple in the Parafilm.
Critical Step
Cut a 200-μl pipette tip with sterile scissors to obtain an opening of 1.5–2 mm in diameter. Be sure that the opening is not too small, as this will disrupt the tissues. Do not attempt to scoop the neuroepithelial tissues out of the well with a spatula or other tools, as this will damage the tissue.
-
12
Remove excess medium from each tissue by carefully sucking off the fluid with an uncut 200-μl tip.
Critical Step
Position the tip with the aggregate behind the opening of the tip to avoid sucking the tissue into the tip, as this will damage the aggregate.
-
13
Immediately add droplets of Matrigel to each aggregate by dripping ∼30 μl onto each tissue so that the droplet fills the Parafilm dimple.
Critical Step
Add the Matrigel quickly to avoid letting the tissues dry out. We typically perform embedding of 16 tissues at a time, a number that is manageable in a time frame that will not cause aggregates to dry out.
-
14
Position each aggregate in the center of the droplet using a 10-μl pipette tip to move the tissue within the droplet (Fig. 2d).
Critical Step
This must be done immediately after adding the droplet, as the Matrigel will begin solidifying once it is at room temperature.
-
15
Place the 60-mm dish containing droplets on Parafilm back into the 37 °C incubator, and incubate it for 20–30 min to allow the Matrigel to polymerize.
-
16
Add 5 ml of cerebral organoid differentiation medium without vitamin A to the 60-mm dish.
-
17
Remove the Matrigel droplets from Parafilm by first using sterile forceps to turn the Parafilm sheet over and by agitating the dish until the droplets fall off the sheet. Any remaining droplets can be removed by using forceps to shake the Parafilm sheet in the medium more vigorously. Continue culturing the tissue droplets in a CO2 incubator.
Stationary culture of expanding neuroepithelial buds
Timing 4 d
-
18
Observe embedded tissues 24 h later under the microscope. Tissues should begin forming buds of more expanded neuroepithelium containing fluid-filled cavities within 1–3 d (Figs. 2e and 3f).
Critical Step
Often, other more migratory cell types will spread from the main mass of tissue into the Matrigel (Fig. 2e). These are typically fibroblast-like cells that most likely represent a small population of non-neural identities that escape the neural induction. However, these cells will typically not survive and their migration away from the tissue seems to promote neuroepithelial bud outgrowth.
-
19
Incubate the droplets for a further 24 h, and then feed the droplets containing neuroepithelial tissues with Cerebral organoid differentiation medium without vitamin A. Incubate it for a further 48 h without agitation.
Critical Step
Change the medium by tilting the dish, allowing the droplets to sink, and by carefully aspirating the medium. Aspirate as much medium as possible without disturbing organoids. Replace it with 5 ml of fresh medium.
Growth of cerebral tissue
Timing up to 1 year
-
20
After 4 d in static culture, transfer the embedded organoids to a 125-ml spinning bioreactor (Fig. 1) by using a cut 1-ml pipette tip with an opening of ∼3 mm. Culture organoids in 75–100 ml of Cerebral organoid differentiation medium containing vitamin A. Place the bioreactor on an appropriate magnetic stir plate installed in the incubator (Fig. 2f). Alternatively, an orbital shaker installed in the incubator, shaking at 85 r.p.m., can be used. Simply replace the medium in each 60-mm dish with cerebral organoid differentiation medium containing vitamin A, and place the dish on the orbital shaker.
Critical Step
Do not transfer more than two 60-mm plates of organoids (32 organoids), as transferring too many organoids to this size flask will lead to fusing of organoids.
Critical Step
Be sure to use a low-speed stir plate approved for use in a tissue culture incubator, as regular stir plates can heat up owing to the magnetic stirring motion.
Critical Step
The use of an orbital shaker enables the analyses of many culture conditions, treatments or genetic variants in parallel, whereas a typical stir plate only includes 4–6 places for separate flasks. We have not observed a difference in morphology of organoids cultured in either the bioreactor or on the orbital shaker.
-
21
Change the medium completely, as described in Step 9, every 3–4 d if organoids are on the shaker or every week for the spinner flask, and monitor for morphology. Perform further analysis on the organoids at your preferred time points, depending on the stage of development desired. See ANTICIPATED RESULTS section for further discussion of when particular stages of development are typically reached. Cryosectioning and immunostaining can be performed as described in Box 1, if desired.
Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
Step 1, making EBs: 1–2 h
Steps 2–4, feeding EBs and initiation of germ layer differentiation: 5–7 d
Steps 5–7, induction of primitive neuroepithelia: 4–5 d
Steps 8–17, transferring neuroepithelial tissues to Matrigel droplets: 1–2 h
Steps 18 and 19, stationary culture of expanding neuroepithelial buds: 4 d
Steps 20 and 21, growth of cerebral tissue: variable; organoids can be maintained for up to 1 year, although tissue growth stops by 2 months and tissues steadily diminish in size after 5–6 months
Box 1, preparation of tissues for analysis and immunostaining: 3 d
Anticipated results
This protocol outlines the generation of 3D cerebral organoid production from hPSCs and their subsequent analysis. The method is easy to implement in a typical tissue culture room using standard equipment. Furthermore, organoids can be examined at various time points to study a variety of developmental stages. By 5–6 d (Step 5), EBs should exhibit optically translucent tissue on the outer surface of the EB (Figs. 2b and 3a). This is typically not variable between EBs within a preparation, although different batches of EBs made at different times can exhibit variability between batches. This variability depends greatly on the morphology and pluripotency of starting PSCs. It is important that PSCs exhibit optimal colony morphology1 (i.e., not >5–10% differentiation and homogenous colonies with obvious borders) before beginning the protocol.
If EBs show proper surface clearing and brightening (Step 5), typically 60–80% of these will exhibit neuroectoderm when placed in neural induction medium (Step 7). However, again batch-to-batch variability can occur, although tissues within a particular batch rarely display much variability at this stage. Finally, neuroepithelial buds will typically form in 30–80% of organoids when placed in Matrigel droplets (Step 18). These buds will be quite heterogeneous from tissue to tissue and will exhibit variable sizes and degrees of continuity. However, large and continuous neuroepithelial tissues will form on many of the organoids if the following precautions are taken to ensure a good batch of organoids: PSCs must exhibit optimal colony morphology before beginning (Figs. 2a); EBs must be transferred to neural induction medium at the correct time according to morphology and size (Figs. 2b and 3a); and tissues must be subsequently transferred to Matrigel droplets at the correct time according to morphology (Figs. 2c and 3c).
Cerebral tissues will generally expand quickly once placed in Matrigel (Figs. 2e and 3f), and by days 15–20 the entire organoid will be difficult to examine using a standard tissue culture microscope as the tissues are too large at this point. Instead, observe the tissues using a dissecting microscope or stereomicroscope to visualize gross morphology. Fluid-filled cavities can often be seen (Fig. 2g), although when located within the tissue mass these are difficult to identify. In addition, retinal regions can be recognized by the presence of pigmented regions reflecting retinal pigmented epithelial identity4.
The large size of organoids after Matrigel embedding necessitates analysis by sectioning and immunohistochemical staining (Box 1). Cryosections performed at an early stage (12–20 d) will reveal expanding neuroepithelium marked by Sox2 or Pax6 expression (Table 2), whereas only occasional neurons are visible13. These neuroepithelia are located adjacent to ventricle-like cavities that are often fluid-filled or contain cellular debris. Occasionally, there may be other tissues exhibiting fibroblast-like morphologies, on the basis of histological characterization, and these are thought to be a side effect of impure neural induction owing to a lack of exogenous growth factors or morphogens. However, these should be a minor population and should not affect growth and development of neural tissues.
After 1 month, organoids should begin to exhibit neuronal differentiation, marked by Tuj1 or DCX expression (Table 2), leading to progressive expansion and thickening of cerebral tissues over the subsequent 1–2 months (Fig. 4a). At this stage, a number of different brain regions are visible (Table 2), including forebrain marked by Foxg1 staining (Fig. 4b) and choroid plexus marked by TTR staining (Fig. 4c), exhibiting a highly convoluted structure and cuboidal epithelial morphology, as well as hippocampus marked by Prox1 and Fzd9 staining (Fig. 4d). Finally, ventral forebrain stains positive for the marker Nkx2.1 (Table 2), whereas retina exhibits retinal layering and pigmented epithelium.

(a) Staining for neurons (TUJ1, green) and progenitors (SOX2, red) in a large continuous cortical tissue within an organoid. Note the organized apical progenitor zone surrounded by basally located neurons. (b) A forebrain region of an organoid staining positive for the marker FOXG1 (red). (c) Choroid plexus stains positive for the marker TTR (green), and it displays convoluted cuboidal epithelium. (d) Hippocampal regions stain positive for the markers PROX1 (green) and FZD9 (red), although the cells fail to spatially organize into recognizable dentate gyrus and CA regions. (e) Staining for mitotic radial glia (P-vimentin (P-vim), green) in a cortical region reveals inner radial glia undergoing mitosis at the apical membrane (arrows), whereas outer radial glia undergo mitosis outside the ventricular zone (arrowheads). All radial glia are marked by SOX2 (red). (f) Staining for cortical layer identities of advanced organoids (75 d). Later-born superficial-layer identity (SATB2, red) neurons populate more superficial regions of the organoid, whereas early-born deep-layer identity (CTIP2, green) neurons populate deeper regions of the organoid. DAPI in a–e labels nuclei (blue). Samples in a–e are 30–35 d after initiation of the protocol. Scale bars, 100 μm (a,b) and 50 μm (c–f).
In addition, cortical regions display evidence of typical progenitor zones, displaying a dense ventricular zone (VZ) populated by Sox2+ inner radial glia (Fig. 4e), whereas outer radial glia, which are also Sox2+, reside outside the VZ. Both populations stain positive for phospho-vimentin during mitosis (Fig. 4e) but divide in different locations: inner radial glia divide at the apical surface, whereas outer radial glia divide outside the VZ. Neurons that are generated migrate outward to form a pre-plate; the precursor to the cortical plate, marked by Reelin and Tbr1 (Table 2), and intermediate progenitors, marked by Tbr2, can be seen in the region adjacent to the VZ.
If allowed to develop further, organoids will progressively produce more neurons, whereas progenitor zones will shrink and eventually disappear by 5–6 months. Tissues thereafter will be composed of primarily fully differentiated neurons, although other populations such as astrocytes and oligodenderocytes have not been examined. The neuron populations present include various layer identities of the cortical plate13,39 (Table 2), which will exhibit rudimentary separation into deep and surface layer distributions (Fig. 4f). However, the neurons will not display a six-layered architecture such as that seen in vivo. Organoids can be further maintained for over 1 year. We have maintained organoids for up to 15 months. Importantly, after 6–7 months, organoids begin shrinking in size, owing to the lack of progenitors and probably to a progressive neuronal cell loss.

References
Lanza, R., Gearhart, J., Hogan, B., Melton, D. & Pedersen, R. Essentials of Stem Cell Biology (Elsevier, 2009).
Sato, T. & Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science 340, 1190–1194 (2013).
Sasai, Y., Eiraku, M. & Suga, H. In vitro organogenesis in three dimensions: self-organising stem cells. Development 139, 4111–4121 (2012).
Eiraku, M. et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472, 51–56 (2011).
Sato, T. et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 459, 262–265 (2009).
Antonica, F. et al. Generation of functional thyroid from embryonic stem cells. Nature 491, 66–71 (2012).
Huch, M. et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 494, 247–250 (2013).
Suga, H. et al. Self-formation of functional adenohypophysis in three-dimensional culture. Nature 480, 57–62 (2011).
Koehler, K.R., Mikosz, A.M., Molosh, A.I., Patel, D. & Hashino, E. Generation of inner ear sensory epithelia from pluripotent stem cells in 3D culture. Nature 500, 217–221 (2013).
Xia, Y. et al. Directed differentiation of human pluripotent cells to ureteric bud kidney progenitor-like cells. Nat. Cell Biol. 15, 1507–1515 (2013).
Takasato, M. et al. Directing human embryonic stem cell differentiation towards a renal lineage generates a self-organizing kidney. Nat. Cell Biol. 16, 118–126 (2014).
Taguchi, A. et al. Redefining the in vivo origin of metanephric nephron progenitors enables generation of complex kidney structures from pluripotent stem cells. Cell Stem Cell 14, 53–67 (2014).
Lancaster, M.A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Nakano, T. et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10, 771–785 (2012).
Spence, J.R. et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470, 105–109 (2011).
Takebe, T. et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 499, 481–484 (2013).
Humphreys, B.D. Kidney structures differentiated from stem cells. Nat. Cell Biol. 16, 19–21 (2013).
Gilbert, S.F. Developmental Biology (Sinauer Associates, 2000).
Evans, M. Discovering pluripotency: 30 years of mouse embryonic stem cells. Nat. Rev. Mol. Cell Biol. 12, 680–686 (2011).
Shevde, N.K. & Mael, A.A. Techniques in embryoid body formation from human pluripotent stem cells. Methods Mol. Biol. 946, 535–546 (2013).
Eiraku, M. et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3, 519–532 (2008).
Zhang, S.C., Wernig, M., Duncan, I.D., Brüstle, O. & Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 19, 1129–1133 (2001).
Hu, B.-Y. & Zhang, S.-C. Directed differentiation of neural-stem cells and subtype-specific neurons from hESCs. Methods Mol. Biol. 636, 123–137 (2010).
Wu, Y., Liu, Y., Chesnut, J.D. & Rao, M.S. Isolation of neural stem and precursor cells from rodent tissue. Methods Mol. Biol. 438, 39–53 (2008).
Price, P.J. & Brewer, G.J. Serum-free media for neural cell cultures. In Protocols for Neural Cell Culture (eds. Federoff, S. and Richardson, A.) 255–264 (Humana Press, 2001).
Elkabetz, Y. et al. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 22, 152–165 (2008).
Gaspard, N. et al. An intrinsic mechanism of corticogenesis from embryonic stem cells. Nature 455, 351–357 (2008).
Brewer, G.J., Torricelli, J.R., Evege, E.K. & Price, P.J. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J. Neurosci. Res. 35, 567–576 (1993).
Koch, P., Opitz, T., Steinbeck, J.A., Ladewig, J. & Brüstle, O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc. Natl. Acad. Sci. USA 106, 3225–3230 (2009).
Siegenthaler, J.A. et al. Retinoic acid from the meninges regulates cortical neuron generation. Cell 139, 597–609 (2009).
Petros, T.J., Tyson, J.A. & Anderson, S.A. Pluripotent stem cells for the study of CNS development. Front. Mol. Neurosci. 4, 30 (2011).
Eiraku, M. & Sasai, Y. Self-formation of layered neural structures in three-dimensional culture of ES cells. Curr. Opin. Neurobiol. 22, 768–777 (2012).
Nasu, M. et al. Robust formation and maintenance of continuous stratified cortical neuroepithelium by laminin-containing matrix in mouse ES cell culture. PLoS ONE 7, e53024 (2012).
Li, M.L. et al. Influence of a reconstituted basement membrane and its components on casein gene expression and secretion in mouse mammary epithelial cells. Proc. Natl. Acad. Sci. USA 84, 136–140 (1987).
Martin, I., Wendt, D. & Heberer, M. The role of bioreactors in tissue engineering. Trends Biotechnol. 22, 80–86 (2004).
Ranga, A., Gjorevski, N. & Lutolf, M.P. Drug discovery through stem cell-based organoid models. Adv. Drug Deliv. Rev. 69–70, 19–28 (2014).
Conti, L. & Cattaneo, E. Neural stem cell systems: physiological players or in vitro entities? Nat. Rev. Neurosci. 11, 176–187 (2010).
Shi, Y., Kirwan, P., Smith, J., Robinson, H.P.C. & Livesey, F.J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 15, 477–86 S1 (2012).
Kadoshima, T. et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 110, 20284–20289 (2013).
Lamonica, B.E., Lui, J.H., Hansen, D.V. & Kriegstein, A.R. Mitotic spindle orientation predicts outer radial glial cell generation in human neocortex. Nat. Commun. 4, 1665 (2013).
Gilmore, E.C. & Walsh, C.A. Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdiscip. Rev. Dev. Biol. 2, 461–478 (2013).
Brennand, K.J. & Gage, F.H. Modeling psychiatric disorders through reprogramming. Dis. Model Mech. 5, 26–32 (2012).
Koike, M., Kurosawa, H. & Amano, Y. A round-bottom 96-well polystyrene plate coated with 2-methacryloyloxyethyl phosphorylcholine as an effective tool for embryoid body formation. Cytotechnology 47, 3–10 (2005).
Acknowledgements
We are grateful to members of the Knoblich laboratory for their technical expertise and feedback, and particularly to M. Renner and A. Peer for their experimental support. We also thank the Stem Cell and BioOptics core facilities of IMBA and the Institute of Molecular Pathology for their technical support. M.A.L. received funding from a European Molecular Biology Organization (EMBO) postdoctoral fellowship, a Helen Hay Whitney postdoctoral fellowship and a Marie Curie International Incoming Fellowship. Work in J.A.K.'s laboratory is supported by the Austrian Academy of Sciences, the Austrian Science Fund (FWF; projects Z153-B09 and I552-B19) and an advanced grant from the European Research Council.
Author information
Authors and Affiliations
Contributions
J.A.K. and M.A.L. wrote the manuscript, and M.A.L. performed the experiments.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Lancaster, M., Knoblich, J. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc 9, 2329–2340 (2014). https://doi.org/10.1038/nprot.2014.158
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2014.158
This article is cited by
-
CUL4B mutations impair human cortical neurogenesis through PP2A-dependent inhibition of AKT and ERK
Cell Death & Disease (2024)
-
Profiling human brain vascular cells using single-cell transcriptomics and organoids
Nature Protocols (2024)
-
Apically localized PANX1 impacts neuroepithelial expansion in human cerebral organoids
Cell Death Discovery (2024)
-
Schizophrenia endothelial cells exhibit higher permeability and altered angiogenesis patterns in patient-derived organoids
Translational Psychiatry (2024)
-
Insights on Three Dimensional Organoid Studies for Stem Cell Therapy in Regenerative Medicine
Stem Cell Reviews and Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
