
Abstract
Adenoviral vectors have shown great promise as vaccine carriers and in gene transfer to correct underlying genetic diseases. Traditionally, cstruction of adenoviral vectors is complex and time consuming. In this paper, we provide an improved method for efficient generation of novel adenoviral vectors by using direct cloning. We introduce a feasible and detailed protocol for the development of chimpanzee adenoviruses (Ads) as molecular clones, as well as for the generation of recombinant virus from the molecular clones. Recombinant viruses are genetically stable and induce potent immune responses in animals. Generation of new Ad molecular clones or new recombinant Ad can be achieved in 2 months or 2 weeks, respectively.
Similar content being viewed by others


Introduction
Vectors based on Ads are commonly used for gene transfer because of their broad tropism, high transduction efficiency and relatively low risk in safety. Until now, most vectors are based on human serotype 5 (AdHu5). This virus is endemic in most human populations, and neutralizing antibodies specific to AdHu5 can be detected in up to 40–60% of humans1. Pre-existing neutralizing antibodies dampen gene transfer efficacy and increase vector-mediated toxicity2. Vectors based on rare human Ad serotypes and Ad from other species are being explored to overcome the impact of pre-existing immunity1,2,3,4,5,6.
Three major methods have traditionally been used to generate recombinant Ad vectors. The most commonly used method is based on homologous recombination in a packaging cell line. In this method, the gene of interest is cloned into a shuttle vector and concomitantly transfected with the Ad genome into HEK 293 cells or other cells that provide E1 in trans. Recombination between the shuttle vector and the Ad genome replaces the E1 domain with the gene of interest. However, recombination is relatively rare and the isolation of recombinant Ad vector using this method is laborious, time consuming and requires multiple rounds of plaque purification7. The second method is based on homologous recombination in Escherichia coli. The commercially available pAdEasy system is based on this method. It is efficient but more complex and requires a three-step transformation, including the use of two different E. coli strains8,9. The third method is based on direct cloning of the Ad genome into a plasmid vector. The genome can then be manipulated in vitro and, following transfection into a packaging cell line, the virus can be rescued. This method is technically challenging because the Ad genome is large (around 36 kb) and contains few useful restriction sites that allow its assembly into a full-length molecular clone8. However, this method is highly desirable, because it is straightforward and it completely eliminates potentially contaminating infectious material from the original Ad genome10 that would not be eliminated by homologous recombination and thus affect the usefulness of Ad vectors for clinical development.
In this paper, we describe a simple strategy that allows the efficient development of Ad molecular clones by direct cloning. Briefly, we took advantage of suitable unique restriction sites within a portion of the genome rather than within the whole genome. Such sites are present in all Ad genomes that we have vectored thus far. Thereafter, we assembled the genome part by part into one plasmid. To clone the foreign gene of interest into the Ad molecular clone, a shuttle vector was used containing two very rare restriction sites, I-CeuI and PI-SceI. The same sites were placed into the molecular clone to allow the insertion of the transgene's expression cassette into the deleted E1 domain. Virus can be rescued by transfection of packaging cell lines with the linearized recombinant molecular clone.
Overview of the protocol
This protocol describes the development of an E1- or E1/E3-deleted Ad molecular clone and the cloning of the gene of interest into the E1/E3-deleted Ad genome, using chimpanzee-derived Ad serotype 6 (AdC6) as an example. The Ad genome contains four segments that encode early gene products (E1–E4), and five segments that encode five late gene products (L1–L5). The E1, E2 and E4 gene products regulate transcription and translation of late genes and are necessary for viral replication. E3 gene products subvert immune responses by altering antigen presentation and cytokine and apoptosis pathways, but are unnecessary for viral replication11. Deletion of the E3 in addition to the E1 domain increases the permitted size of the inserted expression cassette to ∼7.5 kb.
To generate the E1-deleted AdC6 molecular clone, the 5′ right-inverted terminal repeat (ITR) was amplified by PCR and cloned into the pNEB193 vector. Using restriction enzyme sites that are unique in assembly but not necessarily unique to the full AdC6 genome, the right half of the AdC6 genome was then cloned piecemeal into the pNEB193 vector. The left ITR was amplified by PCR and cloned into a different pNEB193 vector. Using the same strategy as above, the remainder of the left fragment of the AdC6 genome was assembled into the pNEB193 vector. Approximately 2.6 kb of the E1 region between SnaBI and NdeI sites was omitted and replaced with a linker that contains the rare enzyme sites of I-CeuI and PI-SceI. This step removes the entire E1a and E1b 19-kDa homolog-coding regions and 74% of the E1b 55-kDa homolog-coding region. Finally, using two suitable enzymes, the E1-deleted left fragment of the Ad genome was released from the pNEB193 vector and inserted into the vector containing the right fragment of the genome, effectively generating the E1-deleted infectious molecular clone of AdC6.
To delete the E3 domain, a fragment including this domain was cut from the E1-deleted molecular clone. The ends of the molecular clone were recombined by ligation. The ∼4 kb E3 region was removed from the fragment using suitable restriction enzymes, i.e., Eco47III and SwaI. The resulting fragment was then cloned back into the E1-deleted molecular clone to generate the E1/E3-deleted Ad molecular clone.
To generate a recombinant molecular clone, first, the gene of interest was cloned into the commercially available pShuttle vector under the control of CMV promoter; other promoters can also be used (see Box 1). The expression cassette was released by digestion with I-CeuI and PI-SceI, and then cloned into the Ad molecular clone that had been cut with the same enzymes. Recombinant Ad vector was generated by rescue in packaging cells such as HEK 293 cells (see Box 2).
This protocol is direct, efficient and reproducible. Provided the sequence of the Ad genome is known, molecular clone generation from a purified Ad averages 2 months and a recombinant Ad vector can be efficiently generated and purified to high titers within an additional 2 weeks. Ads can be cloned as vectors for vaccine development or gene transfer with the use of this protocol. Data obtained from a molecular clone of chimpanzee-derived Ad serotype 7 (AdC7) are also shown.


Results obtained previously using this protocol
Molecular clones from a number of chimpanzee and human Ads have been derived through the use of this protocol. The resulting Ad vectors have undergone extensive testing in vitro. Vectors were genetically stable and efficient expression of the transgene product was maintained through a minimum of 15 passages. Testing in experimental animals showed that vectors induced potent immune responses to a variety of antigens, including those derived from rabies virus1, 12, 13 and 14, influenza virus15, human papilloma virus16,17 and human and simian immunodeficiency viruses18,19,20,21,22,23,24,25,26,27,28.
Materials
REAGENTS
-
EcoRI (New England Biolabs, cat. no. R0101S)
-
KpnI (New England Biolabs, cat. no. R0142S)
-
PmeI (New England Biolabs, cat. no. R0560S)
-
PacI (New England Biolabs, cat. no. R0547S)
-
XbaI (New England Biolabs, cat. no. R0145S)
-
SnaBI (New England Biolabs, cat. no. R0130S)
-
NdeI (New England Biolabs, cat. no. R0111S)
-
SphI (New England Biolabs, cat. no. R0182S)
-
HindIII (New England Biolabs, cat. no. R0104S)
-
AgeI (New England Biolabs, cat. no. R0552S)
-
MluI (New England Biolabs, cat. no. R0198S)
-
SbfI (New England Biolabs, cat. no. R0642S)
-
BstAPI (New England Biolabs, cat. no. V0269S)
-
SwaI (New England Biolabs, cat. no. R0604S)
-
I-CeuI (New England Biolabs, cat. no. R0699S)
-
PI-SceI (New England Biolabs, cat. no. R0696S)
-
SgfI (Promega, cat. no. R5104)
-
Eco47III (Promega, cat. no. R6731)
-
Phusion hot start high-fidelity DNA polymerase (England Biolabs, cat. no. F-540S)
-
dNTP mix (10 mM each; Invitrogen, cat. no. R72501)
-
Alkaline phosphatase (AP; Roche, cat. no. 13826120)
-
T4 DNA ligase (Roche, cat. no. 13827621)
-
DNA ladders (Invitrogen, cat. nos. 10381-010 and 15628-019)
-
Agarose (low melting point; Sigma-Aldrich, cat. no. A9414-25G)
-
UltraPure agarose (Invitrogen, cat. no. 15510-027)
-
l-broth (LB) capsules (MP Biomedicals, cat. no. 3001-031)
-
Ampicillin (Mediatech, cat. no. 61-238-RH)
-
Kanamycin (Roche, cat. no. 106801)
-
Pronase (Boehringer Mannheim, cat. no. 165921)
-
Proteinase K (Boehringer Mannheim, cat. no. 745723)
-
Complete protease inhibitor cocktail tablets (Roche, cat. no. 04693124001)
-
SDS solution (10% (wt/vol); Ambion, cat. no. 9822)
-
Tris-HCl (1 M, pH 8.0; Sigma-Aldrich, cat. no. T-2694)
-
Plasmid DNA mini and midi preparation kit (Qiagen)
-
pShuttle (Clontech, cat. no. K1650-1)
-
pNEB193 (New England Biolabs, cat. no. N3051S)
-
Lipofectamine 2000 transfection reagent (Invitrogen, cat. no. 11668-027)
-
DMEM (Mediatech, cat. no. 10-017-CV)
-
Leibovitz's L-15 medium (L-15; Mediatech, cat. no. 10-045-CV)
-
FBS (PAA Laboratories, cat. no. A11-034)
-
Penicillin-streptomycin (Mediatech, cat. no. 30-002-CI)
-
Trypsin-EDTA (Mediatech, cat. no. 25-053-CI)
-
Primers and oligo linker (Integrated DNA Technologies)
-
Codon-optimized HIV gag (GenScript)
-
DNeasy extraction kit (Qiagen)
-
Distilled water
-
Mice
-
BALB/c mice (ACE Animals)
-
Cells
-
MAX efficiency Stbl2 competent cells (Invitrogen, cat. no. 10268-019)
-
HEK 293 (ATCC, cat. no. CCL-243)
-
Virus
-
AdC6 (Simian Ad23/Pan6; ATCC, cat. no. VR-592)
-
AdC7 (Simian Ad24/Pan7; ATCC, cat no. VR-593)
-
Monoclonal antibody
-
HIV-1 gag (183-H12-5C; NIH, cat. no. 1513)
-
β-Actin (Santa Cruz Biotechnology, cat. no. SC-47778)
-
Buffers
-
Potassium chloride (for KCM buffer; see REAGENT SETUP)
-
Calcium chloride (for KCM buffer; see REAGENT SETUP)
-
Magnesium chloride (for KCM buffer; see REAGENT SETUP)
-
Dulbecco's phosphate-buffered saline (DPBS; Mediatech, cat. no. 21-031-CM)
-
TAE buffer (Mediatech, cat. no. 46-010-CM)
-
RIPA buffer (Pierce, cat. no. 89900)
EQUIPMENT
-
Six-well culture plates
-
T25 tissue culture flasks
-
T175 tissue culture flasks
-
Centrifuges (Eppendorf, cat. nos 5415D and 5810R)
-
Ultracentrifuge (Beckman Coulter)
-
SW 32.1 Ti ultracentrifuge rotor (Beckman Coulter)
-
Ultraclear centrifuge tubes (Beckman, cat. no. 344061)
-
Thermal cycler (Eppendorf Mastercycler gradient, Eppendorf, cat. no. 5331)
-
Water bath
-
Agarose gel chamber
-
Power supply (Bio-Rad)
-
Spectrophotometer
-
Bacteria incubator (37 °C)
-
Humidified tissue culture incubator (37 °C, 5% CO2)
-
Orbital shaker (37 °C)
-
Gel imaging system (Gel logic 200, Kodak)
-
Microscope
-
Freezer (−80 °C)
-
LSRII (Beckman Coulter)
-
Flowjo software 7.1.1 (Treestar)
REAGENT SETUP
Propagate and purify wild-type Ad
-
The wild-type AdC6 was purchased from ATCC, propagated in HEK 293 cells and purified by CsCl gradient centrifugation as described previously29. The number of virus particles (vp) per milliliter was determined by measuring UV absorbance at 260 nm (A260) using a spectrophotometer and was calculated using the following equation30:


KCM buffer (5×)
-
Combine 0.5 M KCl, 0.15 M CaCl2 and 0.25 M MgCl2; buffer can be stored at room temperature (24 °C) for several months.
LB Broth
-
Mix 25 l-broth capsules with 1 liter of distilled water, and then autoclave.
Procedure
Isolation of genomic DNA from wild-type Ad
Timing ∼6 h
-
1
To 100 μl of purified virus (1 × 1013 vp ml−1), add 15 μl of 10% SDS, 15 μl of 100 mg ml−1 pronase, and 3 μl of 20 mg ml−1 proteinase K; then add sterile water to reach a final volume of 150 μl. Incubate for 3 h at 37 °C.
-
2
Use a commercial kit (e.g., DNeasy) to extract genomic DNA of wild-type Ad according to the manufacturer's instructions.
Construction of E1-deleted AdC6 molecular clone
Timing 5 weeks
-
3
Cloning EK (EcoRI to KpnI) fragment of AdC6 into pNEB193 plasmid: digest 0.5 μg of pNEB193 plasmid DNA with KpnI and EcoRI for 2 h at 37 °C. Conduct digestion in a total volume of 30 μl.
-
4
Inactivate the digest for 20 min at 65 °C. Thereafter, add 1 μl of AP for 30 min at 37 °C.
-
5
Aliquot 50 ng of genomic DNA of wild-type AdC6 as template. PCR-amplify the EK fragment using the 5′ and 3′ primers 5′-CACCTCCCGGTACCACATCAC-3′ and 5′-CGGAATTCCGGCGATCGCGCATCATCAATAATATACCTCAAAC-3′. Conduct PCR in a total volume of 50 μl as follows:
Table 2
Phusion HF buffer (5×)
10 μl
(1× Final concentration)
MgCl2 (50 mM)
1 μl
(2.5 mM Total final concentration)
dNTP mix (10 mM)
1 μl
(200 μM Each final concentration)
Forward primer (20 μM)
1 μl
(400 nM Final concentration)
Reverse primer (20 μM)
1 μl
(400 nM Final concentration)
Genomic DNA (50 ng μl−1)
1 μl
(1 ng μl−1 Final concentration)
Phusion polymerase (2 U μl−1)
0.5 μl
(0.02 U μl−1 Final concentration)
Distilled water
34.5 μl
Total volume
50.0 μl
Per reaction
Mix the reaction by vortexing and spin down. Run the PCR for 32 cycles at 94 °C for 1 min, 58 °C for 1 min and at 72 °C for 2.5 min.
-
6
Digest 12 μl of the PCR amplicon with KpnI and EcoRI for 2 h at 37 °C. Conduct digestion in a total volume of 20 μl.
-
7
Run the digestion products on 1% (wt/vol) low-melting agarose gel in TAE buffer. Cut out the bands and place gel slices into Eppendorf tubes. Incubate for 5 min at 68 °C. Cool for 1 min at 37 °C. Set up an in-gel ligation reaction in a total volume of 20 μl, i.e., use 4 μl of vector in liquefied gel, 12 μl of insert in liquefied gel and mix both with 1 μl of 5 U of T4 DNA ligase. Ligate at 16 °C overnight.
Critical Step
Heating the gel slices containing DNA for 5 min at 68 °C is enough to melt the agarose. Try not to exceed 70 °C to avoid denaturing the DNA.
-
8
Melt the ligation products for 5 min at 65 °C. Thereafter, dilute in 180 μl of 1× KCM buffer. Transform 25 μl of diluted ligation product into 50 μl of MAX efficiency Stbl2 competent cells and spread the transformation mix onto an amplicillin-containing LB plate (100 μg ml−1 amplicillin). Incubate plates for 14 h at 30 °C.
-
9
Select five to ten colonies and grow in LB medium for 12–14 h in a shaker at 30 °C and 180 r.p.m. shaking speed. Isolate plasmid DNA by minipreparation. Digest plasmids with the same enzymes used in cloning to check the size of the insert.
Critical Step
Large genomes transformed into MAX efficiency Stb12 competent cells are often unstable. To minimize damage to the DNA, the temperature for bacterial growth in the shaker should not exceed 30 °C, and shaking speed and time should not exceed 200 r.p.m. and 14 h, respectively.
For this and all subsequent cloning steps, confirmation that the desired clone was obtained is made by restriction enzyme digestion, and a fragment of DNA is sequenced to ensure correct ligation. In our example, the new construct was termed PNEB193+EK (Fig. 1). SgfI and EcoRI are sites added to the end of the right ITR of AdC6 by PCR amplification. Both EcoRI and KpnI sites are unique in this part of the genome. A schematic overview of the cloning procedure is shown in Figure 1a.
Figure 1: Flowchart of cloning E1/E3-deleted AdC6 molecular clone. 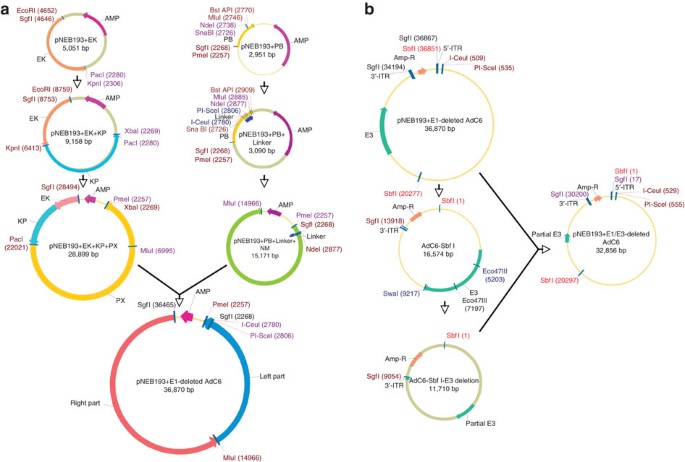
(a) Sequence of cloning E1-deleted AdC6 molecular clone. Details are represented in Steps 3–34. Clones of the right and left halves of wild-type AdC6 were inserted sequentially into two pNEB193 plasmids; PmeI and MluI are attached to the left fragment of AdC6 and to the right fragment of AdC6 in pNEB193 vector, respectively. (b) Sequential illustration of cloning E1/E3-deleted AdC6 molecular clone. The procedure is detailed in Steps 35–44. In brief, one fragment including E3 domain was cut from E1-deleted molecular clone, E3 region was then removed from the fragment by digestion of Eco47III and SwaI. Finally, the resulting fragment was cloned back into E1-deleted molecular clone.
-
10
Cloning of the KP (KpnI to PacI) fragment of AdC6 into the PNEB193+EK plasmid: digest 0.5 μg of PNEB193+EK plasmid DNA with PacI and KpnI. Conduct the reaction in a total volume of 30 μl and incubate for 3 h at 37 °C.
-
11
Repeat Step 4.
-
12
Digest 1 μg of genomic DNA of wild-type AdC6 with PacI and KpnI to release the KP fragment from AdC6. Conduct the reaction in a total volume of 20 μl and incubate the mixture for 3 h at 37 °C.
-
13
Repeat Steps 7–9. Both KpnI and PacI sites are unique in this part of the genome. The new construct was termed PNEB193+EK+KP.
-
14
Cloning of the PX (PacI to XbaI) fragment of AdC6 into the PNEB193+EK+KP plasmid: digest 0.5 μg of PNEB193+EK+KP plasmid DNA with XbaI and PacI. Conduct the reaction in a total volume of 30 μl and incubate for 3 h at 37 °C.
-
15
Repeat Step 4.
-
16
Digest 1 μg of genomic DNA of wild-type AdC6 with XbaI and PacI to release the XP fragment. Conduct the reaction in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
17
Repeat Steps 7–9. Both PacI and XbaI sites are unique in this part of the genome. The new construct was termed PNEB193+EK+KP+PX.
-
18
Cloning the PB (PmeI to BstAPI) fragment of AdC6 into the PNEB193 plasmid: digest 0.5 μg of pNEB293 plasmid DNA with PmeI for 3 h at 37 °C, then add BstAPI at 1.5 μl for 3 h at 60 °C. Conduct digestion in a total volume of 30 μl.
-
19
Inactivate the digest for 20 min at 80 °C; add 1 μl of AP for 30 min at 37 °C.
-
20
Use 50 ng of genomic DNA of wild-type AdC6 as template, amplify PB fragment by PCR for introduction of the PmeI and SgfI sites to the end of the left ITR of AdC6 and introduce NdeI, MluI and BstAPI at the right end of this fragment. The following 5′ and 3′ primers were used: 5′-GGGTTTAAACCCGCGATCGCGCATCATCAATAATATACCTCAAAC-3′ and 5′-AAGGGCACCATCTGCACGCGTCGCCATATGGAATACGTAAAAACACCGGACTTTG-3′. Conduct the PCR reaction in a total volume of 50 μl as follows.
Table 3
Phusion HF buffer (5×)
10 μl
(1× Final concentration)
MgCl2 (50 mM)
1 μl
(2.5 mM Total final concentration)
dNTP mix (10 mM)
1 μl
(200 μM Each final concentration)
Forward primer (20 μM)
1 μl
(400 nM Final concentration)
Reverse primer (20 μM)
1 μl
(400 nM Final concentration)
Genomic DNA (50 ng μl−1)
1 μl
(1 ng μl−1 Final concentration)
Phusion polymerase (2 U μl−1)
0.5 μl
(0.02 U μl−1 Final concentration)
Distilled water
34.5 μl
Total volume
50.0 μl
Per reaction
-
21
Vortex and spin down all the fluids to obtain a pellet. Run PCR for 32 cycles at 94 °C for 1 min, 62 °C for 50 s and 72 °C for 40 s.
-
22
Digest 12 μl of PCR amplicon with PmeI for 3 h at 37 °C; thereafter, add 1.5 μl of BstAPI and incubate for 3 h at 60 °C. Conduct digestion in a total volume of 30 μl.
Repeat Steps 7–9. Both PmeI and BstAPI sites are unique in this part of the genome. The new construct was termed PNEB193+PB.
-
23
Cloning of the linker into the PNEB193+PB plasmid: digest 0.5 μg of PNEB193+PB plasmid DNA with SnaBI and NdeI. These two enzyme sites are unique in this plasmid. Conduct the reaction in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
24
Repeat Step 4.
-
25
A linker with a length of 165 bp containing the extremely rare enzymes I-CeuI and PI-SceI was synthesized by Integrated DNA Technologies. The sequence of the linker is as follows:
5′-cgctacgtacgatatcatttccccgaaagtgccacctgaccgtaactataacggtcctaaggtagcgaagcatctatgtcgggt‐gcggagaaagaggtaatgaaatggcattatgggtattatgggtctgcattaatgaatcggtcagatatcgacatatgctgc-3′.
Digest 0.3 μg of the linker with SnaBI and NdeI. Conduct the reaction in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
26
Repeat Steps 7–9. The new construct was termed PNEB193+PB+Linker.
-
27
Cloning NM (NdeI to MluI) fragment into PNEB193+PB+Linker plasmid: digest 0.5 μg of the PNEB193+PB+Linker plasmid with NdeI and MluI. Conduct the reaction in a total volume of 30 μl and incubate for 3 h at 37 °C.
-
28
Repeat Step 4.
-
29
Digest 1 μg of genomic DNA of wild-type AdC6 with NdeI and MluI to release the NM fragment. Conduct the reaction in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
30
Repeat Steps 7–9. Both NdeI and MluI sites are unique in this fragment of the genome. This new construct was termed PNEB193+PB+Linker+NM.
-
31
Construct the full-length E1-deleted AdC6 molecular clone: digest 0.5 μg of PNEB193+EK+KP+PX plasmid with PmeI and MluI. Conduct the reaction in a total volume of 30 μl. Incubate for 3 h at 37 °C.
-
32
Repeat Step 4.
-
33
Digest 0.5 μg of PNEB193+PB+Linker+NM with PmeI and MluI to release the fragment of PB+Linker+NM.
-
34
Repeat Steps 7–9. Both PmeI and MluI sites are unique in the genome of AdC6. The new construct represents an infectious clone of the E1-deleted AdC6 within a PNEB193 vector. It was termed AdC6–000.

Generate E1/E3-deleted AdC6 molecular clone
Timing ∼10 d
-
35
Digest 0.5 μg of AdC6-000 plasmid with SbfI. Conduct the reaction in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
36
Run the digest on 1% (wt/vol) low-melting agarose gel in TAE buffer; cut out a band containing a fragment of ∼19 kb and place it into an Eppendorf tube. Incubate for 5 min at 68 °C. Cool for 1 min at 37 °C. Assemble an in-gel ligation reaction as described in Step 7.
-
37
Repeat Steps 8 and 9. The ligation results in the new construct AdC6-Sbf1.
-
38
Digest 0.5 μg of AdC6-Sbf I plasmid DNA with Eco47III and SwaI. Both of these enzyme sites are unique in this plasmid and both enzymes produce blunt ends. Conduct the reaction in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
39
Run all digests on 1% (wt/vol) low-melting agarose gels in TAE buffer; cut out a band containing a fragment of ∼15 kb and place it into an Eppendorf tube. Heat for 5 min at 68 °C. Cool down for 1 min at 37 °C. Conduct an in-gel ligation as described in Step 7.
-
40
Repeat Steps 8 and 9. The ligation results in the new construct AdC6-Sbf1-E3 deletion.
-
41
Digest 0.5 μg of AdC6-000 plasmid with SbfI. Conduct the reaction in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
42
Inactivate the digest for 20 min at 65 °C. Thereafter, add 1 μl of AP for 30 min at 37 °C.
-
43
Digest AdC6-Sbf1-E3 deletion with SbfI. Perform the reaction in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
44
Repeat Steps 7–9. The new construct is the E1/E3-deleted AdC6 molecular clone, and was termed AdC6 010. A schematic overview of the cloning procedure is shown in Figure 1b.
Clone foreign gene into the AdC6 010 vector
Timing ∼1 week
-
45
Cloning of the HIV gag gene into pShuttle: digest 0.5 μg of pShuttle plasmid DNA with KpnI and XbaI for 3 h at 37 °C. Conduct digestion in a total volume of 20 μl.
-
46
Repeat Step 4.
-
47
Codon-optimized HIV gag was synthesized by GenScript and provided in pUC57 vector flanked by XbaI and KpnI sites. Digest 0.5 μg of pUC57-HIV gag with KpnI and XbaI. Conduct digestion in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
48
Repeat Steps 7–9 to generate pShuttle-HIV gag.
Critical Step
pShuttle contains kanamycin resistance gene. The transformation mixture should be spread onto a kanamycin-containing LB plate (50 μg ml−1 of kanamycin).
-
49
Cloning of the HIV gag gene into the AdC6 010 vector: digest 0.5 μg of the AdC6 010 plasmid with I-CeuI and PI-SceI. Conduct the reaction in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
50
Repeat Step 4.
-
51
Digest 0.5 μg of pShuttle-HIV gag plasmid with I-CeuI and PI-SceI. Conduct the reaction in a total volume of 20 μl and incubate for 3 h at 37 °C.
-
52
Repeat Steps 7–9. The new construct was termed AdC6 010-HIV gag. A schematic overview of the cloning procedure is shown in Figure 2.
Figure 2: Schematic representation of the cloning of the gene of interest into E1/E3-deleted AdC6 and virus production. 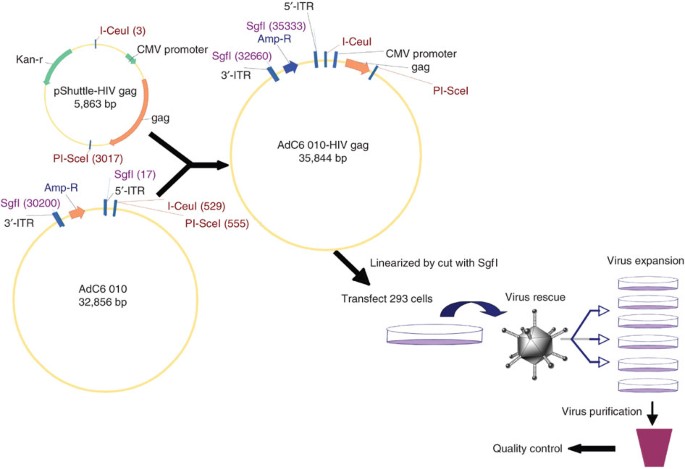
Foreign gene of interest was cloned into pShuttle and then into E1/E3-deleted AdC6 molecular clone (for detailed procedures refer to Steps 45–52). Recombinant molecular clone was then transfected into HEK 293 cells after linearization. Virus was expanded and purified after rescue (refer to Steps 53–61).

Transfection of HEK 293 cells with the linearized AdC6 010-HIV gag construct and rescue of virus
Timing 6–8 d
-
53
Digest 5 μg of AdC6 010-HIV gag with SgfI to linearize the plasmid. Conduct the reaction in a total volume of 50 μl and incubate for 3 h at 37 °C.
-
54
Inactivate the digestion mixture for 20 min at 65 °C.
-
55
Combine the inactivated digestion mixture with Lipofectamine 2000 transfection reagent; transfect one T25 flask of HEK293 cells at a confluency of 80–90% following the manufacturer's instruction.
-
56
At 4 h after transfection, substitute the medium with DMEM containing 5% FBS and incubate at 37 °C. Virus plaques become visible 6–8 d later. Substitute the medium if necesssary.
Virus amplification in HEK 293 cells
Timing ∼10 d
-
57
Harvest transfected HEK 293 cells once virus plaques are readily detectable, and resuspend in 1 ml of L15 medium. Freeze and thaw cells three times. Spin the lysate for 8 min at 4,000 r.p.m. at 4 °C.
-
58
Use half of the resulting supernatant to infect one T175 flask of HEK 293 cells at a confluence of 90%. Reserve the remainder of the supernatant at −80 °C. After 24–48 h, once viral plaques become visible, harvest infected cells and resuspend in 1 ml of L15 medium. Repeat the above freeze-and-thaw procedure and save the supernatant.
-
59
Use three-fourths of the supernatant to infect four T175 flasks of HEK 293 cells at a confluence of 90%. After 24 h, harvest infected cells, and resuspend in 1 ml of L15 medium. Repeat the above freeze-and-thaw procedure and save the supernatant.
-
60
Use three-fourths of the supernatant, infect 40 T175 flasks of HEK 293 cells at a confluence of 90%. After 24 h, harvest infected cells, and resuspend in 30 ml of 10 mM Tris-HCl buffer. Repeat the above freeze-and-thaw procedure and save the supernatant.
-
61
Purify the virus from the supernatant by CsCl gradient centrifugation, determine the number of vps by spectrophotometry and determine virus infectivity by titration on HEK 293 cells31.
Testing genetic stability of the vector
Timing 3–4 weeks
-
62
Repeat Step 58 to passage the virus serially 15 times (p15). Thereafter, repeat Steps 59–61 to expand and purify the p15 virus.
-
63
Repeat Steps 1 and 2 to isolate the genomic DNA of the p15 virus. Digest 1.5 μg of p15 genomic DNA with SphI, HindIII and AgeI. Conduct the reactions in a total volume of 20 μl and incubate for 3 h at 37 °C. Use identical digestions for viral DNA from the first passage virus.
-
64
Run all digests in 1% (wt/vol) agarose gel to determine whether they have the correct fragments (shown in Fig. 3a).
Figure 3: Genetic stability test and the expression of adenoviral vectors. 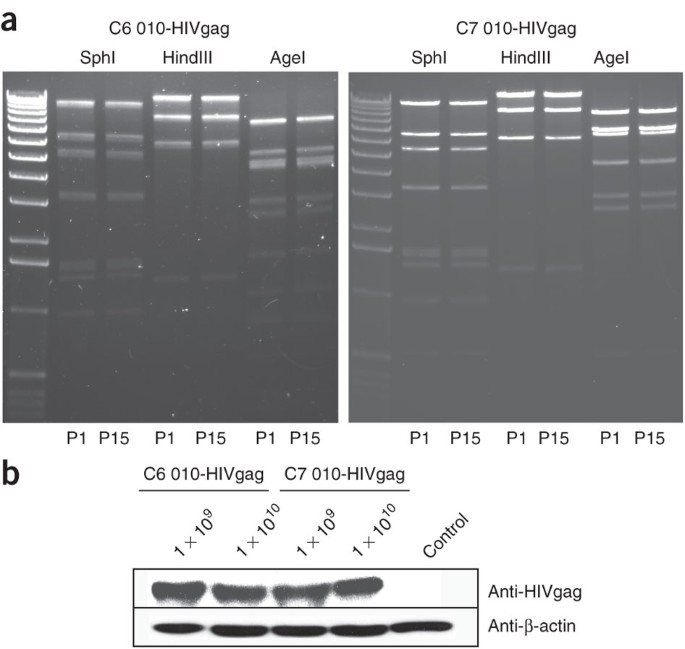
(a) Genomic DNAs from passage 15 of the recombinant adenovirus were purified and then digested with SphI, HindIII and AgeI. The same digestions for the virus DNA from the first passage were set in parallel. Digests were run on 1% (wt/vol) agarose gel. (b) HEK 293 cells were infected with 1 × 109 and 1 × 1010 vps of AdC6 010-HIV gag or AdC7 010-HIV gag. After 24 h, cells were harvested and lysed. Total protein of each sample was run on 10% SDS–polyacrylamide gel electrophoresis gel. The expression of HIV gag was detected by blotting with anti-HIV Gag antibody; antibody to β-actin was included.

Expression of the transgene product
Timing 4 d
-
65
Seed HEK 293 cells in a six-well plate at a density that results in 90% confluency within 24 h.
-
66
The next day, infect the cells in separate wells with 1 × 109 or 1 × 1010 vps of recombinant Ad vector.
-
67
After 24 h, harvest cells and resuspend in 100 μl of RIPA buffer with protease inhibitors.
-
68
Run the western blot to detect the expression of HIV gag by blotting with an anti–HIV Gag antibody. β-Actin is included as a loading control (Fig. 3b).
Testing the immune responses induced by Ad vectors in mice
Timing 2–3 weeks
-
69
Immunize female BALB/c mice (6–8 weeks old) with either AdC6 010-HIV gag or AdC7 010-HIV gag dosed at 1 × 108, 1 × 109 or 1 × 1010 vps.
-
70
After 2 weeks, obtain blood samples from the mice and determine frequencies of HIV gag–specific CD8+ T cells by staining peripheral blood mononuclear cells with fluorochrome-labeled antibodies to CD8 and a MHC class I tetramer specific for the HIV Gag immunodominant epitope (AMQMLKETI) as described32.
-
71
Analyze stained cells on an LSRII (Beckman Coulter) and conduct postacquisition analysis with Flowjo software 7.1.1 (Treestar).
Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
Steps 1 and 2, Isolate genomic DNA of wild-type Ad: 6 h
Steps 3–34, Generate E1-deleted AdC6 molecular clone: ∼5 weeks
Steps 35–44, Generate E1/E3-deleted AdC6 molecular clone: ∼10 d
Steps 45–52, Clone gene of interest into E1/E3-deleted AdC6 molecular clone: ∼1 week
Steps 53–56, Rescue virus from packing cell line: 6–8 d
Steps 57–61, Expand and purify virus: ∼10 d
Steps 62–64, Test genetic stability: ∼3–4 weeks
Steps 65–68, Test transgene expression: 4 d
Steps 69–71, Test immune responses in mice: 2–3 weeks
Anticipated results
The virus yields for AdC6 010-HIV gag and AdC7 010-HIV gag preparation from cultures of adherent HEK 293 cells (40 T175 flasks) should be greater than 2 × 1013 vps, with a vp–infectious unit ratio below 200 (see Box 3). Alternative methods based on amplification of Ad vectors in suspension cultures have been described previously33. Viruses should remain stable on serial passages. After immunization, vectors should elicit readily detectable transgene product-specific CD8+ T-cell responses (Fig. 4).

Groups of BALB/c mice were immunized i.m. with 1 × 108, 1 × 109 and 1 × 1010 vps of AdC6 010-HIV gag or AdC7 010-HIV gag. Peripheral blood mononuclear cells of individual mice were analyzed 14 d later by tetramer staining and for cell surface-expressed CD8 and tetramer HIV gag. Graph shows mean percentages of CD8+ Gag tetramer cells/CD8+ cells from individual mice ± s.d.
Depending on the transgene product, other methods may have to be used to test for immune responses. Transgene products designed to induce B-cell responses should be tested for induction of antibodies from serum or plasma by suitable assays such as neutralization assays for viral glycoproteins or enzyme-linked immunosorbent assays for antigens that fail to elicit neutralizing antibodies. B-cell responses can be enumerated by ELISpot assays for antibody-secreting cells or memory B cells34. Induction of antibody responses can be assessed in inbred or outbred strains of mice. Induction of T cells can be tested for by a variety of assays if T-cell epitopes or their restricting elements are not defined; the definition of both is a prerequisite for the development of tetramers. Alternative T cell assays include intracellular cytokine staining upon a short in vitro stimulation of lymphocytes with their cognate antigen; ELISpot assays, which do not distinguish between CD8+ and CD4+ T-cell subsets, unless one of these subsets is depleted from the lymphocyte preparation before analyses; and others such as 51Cr-release or cytokine-release assays, which assess function rather than magnitude of a T-cell response. It should be noted that Ad vectors induce preferentially potent CD8+ T-cell responses, whereas induction of CD4+ T-cell responses is less prominent. Attention should be paid to the presence of a suitable T-cell epitope within the transgene product when selecting a mouse strain for immunogenicity testing of Ad vectors. In cases in which the transgene product lacks an epitope that can be recognized by T cells of any of the available inbred mouse strains, testing may have to be conducted in another species or in HLA transgenic mice35.
The timeline described above is based on our experience with AdC6. A number of problems can be encountered during the procedures, and this can significantly expand the time line. Problems we have encountered in the past included contamination of wild-type Ad virus with variants, which required repeated cloning of the virus by plaque purification (add ∼4 weeks); inaccurate sequence information, which did not identify all of the sites for restriction enzymes we had planned to use (add anywhere from 1 to 8 weeks if the viral genome has to be resequenced or if a new cloning strategy has to be developed and implemented); problems with ligations (add ∼3–6 d); and, most worrisome, problems with viral rescue. We typically try at first to rescue a molecular clone that does not carry a transgene. If virus cannot be rescued, it commonly means that the molecular clone is flawed, which can best be detected by sequencing of key regions (ITRs and regions that could have been modified during cloning).

References
Xiang, Z. et al. Novel, chimpanzee serotype 68-based adenoviral vaccine carrier for induction of antibodies to a transgene product. J. Virol. 76, 2667–2675 (2002).
Peruzzi, D. et al. A novel chimpanzee serotype-based adenoviral vector as delivery tool for cancer vaccines. Vaccine 27, 1293–1300 (2009).
Liu, J. et al. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature 7225, 87–91 (2009).
Roy, S. et al. Characterization of a family of chimpanzee adenoviruses and development of molecular clones for gene transfer vectors. Hum. Gene Ther. 15, 519–530 (2004).
Seshidhar, R.P. et al. Development of adenovirus serotype 35 as a gene transfer vector. Virology 311, 384–393 (2003).
Roy, S. et al. Generation of an adenoviral vaccine vector based on simian adenovirus 21. J. Gen. Virol. 87, 2477–2485 (2006).
Davis, A.R., Wivel, N.A., Palladino, J.L., Tao, L. & Wilson, J.M. Construction of adenoviral vectors. Methods Mol. Biol. 135, 515–523 (2000).
Mizuguchi, H. & Kay, M.A. Efficient construction of a recombinant adenovirus vector by an improved in vitro ligation method. Hum. Gene Ther. 9, 2577–2583 (1998).
Danthinne, X. & Werth, E. New tools for the generation of E1-and/or E3-substituted adenoviral vectors. Gene Ther. 7, 80–87 (2000).
Gao, G. et al. High throughput creation of recombinant adenovirus vectors by direct cloning, green-white selection and I-Sce I-mediated rescue of circular adenovirus plasmids in 293 cells. Gene Ther. 22, 1926–1930 (2003).
Lasaro, M.O. & Ertl, H.C. New insights on adenovirus as vaccine vectors. Mol. Ther. 17, 1333–1339 (2009).
Xiang, Z.Q., Gao, G.P., Reyes-Sandoval, A., Li, Y., Wilson, J.M. & Ertl, H.C. Oral vaccination of mice with adenoviral vectors is not impaired by preexisting immunity to the vaccine carrier. J. Virol. 77, 10780–10789 (2003).
Xiang, Z., Li, Y., Gao, G., Wilson, J.M. & Ertl, H.C. Mucosally delivered E1-deleted adenoviral vaccine carriers induce transgene product-specific antibody responses in neonatal mice. J. Immunol. 171, 4287–4293 (2003).
Zhou, D., Cun, A., Li, Y., Xiang, Z. & Ertl, H.C. A chimpanzee-origin adenovirus vector expressing the rabies virus glycoprotein as an oral vaccine against inhalation infection with rabies virus. Mol. Ther. 14, 662–672 (2006).
DiMenna, L.J. et al. Augmentation of primary influenza A virus specific CD8+ T cell responses in aged mice through blockade of an immunoinhibitory pathway. J. Immunol. 184, 5475–5484 (2010).
Lasaro, M.O. et al. Targeting of antigen to the herpesvirus entry mediator augments primary adaptive immune responses. Nat. Med. 14, 205–212 (2008).
He, Z. et al. Viral recombinant vaccines to the E6 and E7 antigens of HPV-16. Virology 270, 146–161 (2000).
Fitzgerald, J.C. et al. A simian replication-defective adenoviral recombinant vaccine to HIV-1 gag. J. Immunol. 170, 1416–1422 (2003).
Pinto, A.R., Fitzgerald, J.C., Giles-Davis, W., Gao, G.P., Wilson, J.M. & Ertl, H.C. Induction of CD8+ T cells to an HIV-1 antigen through a prime boost regimen with heterologous E1-deleted adenoviral vaccine carriers. J. Immunol. 171, 6774–6779 (2003).
Pinto, A.R., Fitzgerald, J.C., Gao, G.P., Wilson, J.M. & Ertl, H.C. Induction of CD8+ T cells to an HIV-1 antigen upon oral immunization of mice with a simian E1-deleted adenoviral vector. Vaccine 22, 697–703 (2004).
Reyes-Sandoval, A. et al. Human immunodeficiency virus type 1-specific immune responses in primates upon sequential immunization with adenoviral vaccine carriers of human and simian serotypes. J. Virol. 78, 7392–739 (2004).
Tatsis, N. et al. Chimpanzee-origin adenovirus vectors as vaccine carriers. Gene Ther. 3, 421–429 (2006).
Tatsis, N. et al. A CD46-binding chimpanzee adenovirus vector as a vaccine carrier. Mol. Ther. 15, 608–617 (2007).
Lin, S.W., Cun, A.S., Harris-McCoy, K. & Ertl, H.C. Intramuscular rather than oral administration of replication-defective adenoviral vaccine vector induces specific CD8+ T cell responses in the gut. Vaccine 25, 2187–2193 (2007).
McCoy, K. et al. Effect of preexisting immunity to adenovirus human serotype 5 antigens on the immune responses of nonhuman primates to vaccine regimens based on human- or chimpanzee-derived adenovirus vectors. J. Virol. 81, 6594–6604 (2007).
Tatsis, N. et al. Adenoviral vectors persist in vivo and maintain activated CD8+ T cells: implications for their use as vaccines. Blood 110, 1916–1923 (2007).
Tatsis, N., Lin, S.W., Harris-McCoy, K., Garber, D.A., Feinberg, M.B. & Ertl, H.C. Multiple immunizations with adenovirus and MVA vectors improve CD8+ T cell functionality and mucosal homing. Virology 367, 156–167 (2007).
Tatsis, N. et al. Adenovirus vector-induced immune responses in nonhuman primates: responses to prime boost regimens. J. Immunol. 182, 6587–6599 (2009).
Green, A.P. et al. A new scalable method for the purification of recombinant adenovirus vectors. Hum. Gene Ther. 13, 1921–1934 (2002).
Bastaki, M., Braiterman, L.T., Johns, D.C., Chen, Y.H. & Hubbard, A.L. Absence of direct delivery for single transmembrane apical proteins or their 'secretory' forms in polarized hepatic cells. Mol. Biol. Cell. 13, 225–237 (2002).
Varnavski, A.N., Calcedo, R., Bove, M., Gao, G. & Wilson, J.M. Evaluation of toxicity from high-dose systemic administration of recombinant adenovirus vector in vector-naive and pre-immunized mice. Gene Ther. 12, 427–436 (2005).
Lin, S.W., Hensley, S.E., Tatsis, N., Lasaro, M.O. & Ertl, H.C. Recombinant adeno-associated virus vectors induce functionally impaired transgene product-specific CD8+ T cells in mice. J. Clin. Invest. 117, 3958–3970 (2007).
Joo, H.M., He, Y. & Sangster, M.Y. Broad dispersion and lung localization of virus-specific memory B cells induced by influenza pneumonia. Proc. Natl. Acad. Sci. USA 105, 3485–3490 (2008).
Taneja, V. & David, C.S. HLA transgenic mice as humanized mouse models of disease and immunity. J. Clin. Invest. 101, 921–926 (1998).
Cortin, V., Thibault, J., Jacob, D. & Garnier, A. High-titer adenovirus vector production in 293S cell perfusion culture. Biotechnol. Prog. 20, 858–863 (2004).
Stanton, R.J., McSharry, B.P., Armstrong, M., Tomasec, P. & Wilkinson, G.W. Re-engineering adenovirus vector systems to enable high-throughput analyses of gene function. Biotechniques 45, 659–662, 664–668 (2008).
Sakhuja, K., Reddy, P.S., Ganesh, S., Cantaniag, F., Pattison, S. & Limbach, P. et al. Optimization of the generation and propagation of gutless adenoviral vectors. Hum. Gene Ther. 14, 243–54 (2003).
Abbink, P., Lemckert, A.A., Ewald, B.A., Lynch, D.M., Denholtz, M. & Smits, S. et al. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 81, 4654–4663 (2007).
Limbach, K.L. & Richie, T.L. Viral vectors in malaria vaccine development. Parasite Immunol. 31, 501–509 (2009).
Acknowledgements
This work was funded by grants from NIAID and the Commonwealth of Pennsylvania. We thank C. Cole for her help in preparing the paper.
Author information
Authors and Affiliations
Contributions
D.Z. designed and performed experiments, analyzed data and wrote the paper. X.Z. designed and performed experiments and analyzed data. A.B., H.L., H.C., J.C.S., Y.L., W.G.-D. and Z.X. performed experiments and analyzed data. H.C.J.E. supervised all experimental procedures and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Zhou, D., Zhou, X., Bian, A. et al. An efficient method of directly cloning chimpanzee adenovirus as a vaccine vector. Nat Protoc 5, 1775–1785 (2010). https://doi.org/10.1038/nprot.2010.134
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2010.134
This article is cited by
-
Intranasal multivalent adenoviral-vectored vaccine protects against replicating and dormant M.tb in conventional and humanized mice
npj Vaccines (2023)
-
Hepatitis B virus polymerase-specific T cell epitopes shift in a mouse model of chronic infection
Virology Journal (2021)
-
Prime-boost vaccination with recombinant protein and adenovirus-vector expressing Plasmodium vivax circumsporozoite protein (CSP) partially protects mice against Pb/Pv sporozoite challenge
Scientific Reports (2018)
-
Methods and clinical development of adenovirus-vectored vaccines against mucosal pathogens
Molecular Therapy - Methods & Clinical Development (2016)
-
Hemagglutinin-targeting Artificial MicroRNAs Expressed by Adenovirus Protect Mice From Different Clades of H5N1 Infection
Molecular Therapy - Nucleic Acids (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
