
Abstract
This protocol describes the application of fluorescence in situ hybridization (FISH) to three-dimensionally (3D) preserved tissue sections derived from intact plant structures such as roots or florets. The method is based on the combination of vibratome sectioning with confocal microscopy. The protocol provides an excellent tool to investigate chromosome organization in plant nuclei in all cell types and has been used on tissues of both monocot and dicot plant species. The visualization of 3D well-preserved tissues means that cell types can be confidently identified. For example, meiocytes can be clearly identified at all stages of meiosis and can be imaged in the context of their surrounding maternal tissue. FISH can be used to localize centromeres, telomeres, repetitive regions as well as unique regions, and total genomic DNAs can be used as probes to visualize chromosomes or chromosome segments. The method can be adapted to RNA FISH and can be combined with immunofluorescence labeling. Once the desired plant material is sectioned, which depends on the number of samples, the protocol that we present here can be carried out within 3 d.
Similar content being viewed by others

Introduction
Many of the most interesting aspects of plant cell biology and development occur in cells deep within tissues of the plant. Examples are male meiosis occurring within the anthers, and embryogenesis and endosperm development occurring within the developing seed. These cells are difficult or impossible to image, even within confocal microscopy, which is rarely able to image deeper than 100 μm. Thus to be accessible to imaging, dissection or sectioning must be used. Vibratome tissue sectioning is a simple way to produce relatively thick sections (20–50 μm), which can be imaged to reveal the structure of the underlying tissues. It has the advantage of preserving 3D structures well, so that subcellular organization can be reliably imaged. Furthermore, reliable identification of cell types often requires an accurate assessment of the tissue context, which is lost when cytological squash preparations are made.
This method does not require embedding of tissue in wax or resin, and can be applied to fixed or unfixed tissues. The only requirements are that the plant tissue has to be sufficiently rigid for sectioning, and the plant parts have to be large enough to handle in the vibratome. These requirements are met by the roots and florets and other organs of many plants. We have used it for both roots and florets of wheat (Triticum aestivum L.) and related species1,2,3, for rye (Secale cereale L.) florets4, for rice (Oryza sativa L.) roots and florets5 and maize (Zea mays L.) roots6 among the cereals, and for roots of pea (Pisum sativum L.), bean (Vicia faba L.)7 and soya (Glycine max L.)8. The only plant species with which we have been unsuccessful in vibratome sectioning is Arabidopsis thaliana, because of its small size. By carrying out the sectioning in 100% ethanol, we have also been able to apply vibratome sectioning to early-stage wheat endosperm9. The resulting vibratome sections can be used in many different types of labeling experiments. We have used such sections for DNA FISH, for RNA FISH to detect transcripts10, for antibody immunofluorescence labeling8 and for labeling of nascent transcripts by BrUTP incorporation in unfixed tissue. We have also combined antibody and BrUTP labeling with FISH (e.g., ref. 11).
This type of sectioning combined with in situ hybridization (ISH) provides good specificity and resolution for screening large number of plants in the required developmental stages, and allows reliable and statistically significant data sets to be collected with a reasonable expenditure of time and effort. FISH and genomic in situ hybridization (GISH) are now well-established techniques for cytogenetical analyses and several groups have successfully applied these methods to wheat and its relatives (e.g. ref. 12). The physical size of introgressed segments can be assessed by GISH, which can distinguish between related genomes when total genomic DNA is used as a probe13, and the chromosomal distribution of repetitive sequences can be studied14,15.
We have used this protocol extensively to investigate plant nuclear organization in cereals, which provide excellent models for studying chromosome behavior in a range of tissue types. It provides a reliable tool to answer questions about the nature and timing of recognition events, chromosome associations and control mechanisms before and during meiosis, and to determine, for example, how universal meiotic events are in plants over a wide range of genome sizes and taxa2,3,5,15,16.
Materials
Reagents
-
Plant material of interest (we have successfully used wheat, rye, rice, maize, pea, bean and soya)
-
Multiwell slides (ICN Biomedicals Inc.)
-
Decon 90 (Decon Laboratories Limited)
-
3-Aminopropyl triethoxy silane (APTES; Sigma)
-
Acetone (Merck)
-
Glutaraldehyde (Merck)
-
1× PBS buffer pH 7 (150 mM NaCl, 10 mM sodium phosphate pH 7.5, 1 mM EDTA)
-
100 mM DTT (Promega P117B)
-
100 mM dNTP (Roche 11 969 064 001)
-
Biotin-16-dUTP (Roche 11 093 070 910)
-
Digoxigenin-11-dUTP (Roche 11 093 088 910)
-
DNA polI/DNaseI (Invitrogen 18162-016)
-
0.5 M EDTA pH 8
-
MinElute PCR purification kit (Qiagen 28004)
-
Hybond N+ membrane (Amersham Biosciences RPN303B)
-
1 M Tris-HCl pH 7.5
-
1 M Tris-HCl pH 9.5
-
3 M NaCl
-
1 M MgCl2
-
Blocking reagent (Roche 1096176)
-
Anti-digoxigenin-AP Fab fragment (Roche 11 093 274 910)
-
Anti-biotin-AP Fab fragment (Roche 11426303001)
-
4-Nitrobluetetrazolium chloride (NBT; Roche 11 383 213 001)
-
5-Bromo 4-chloro-3-indolyl-phosphate (BCIP; Roche 11 383 221 001)
-
Fixative: 4% formaldehyde (wt/vol) in 1× PEM buffer (50 mM PIPES/KOH pH 6.5, 5 mM EGTA, 5 mM MgSO4)
-
Methanol: 30%, 50%, 70% and 100% (vol/vol)
-
Cellulase (Onozuka R-10)
-
Pectolyase (Kikkoman)
-
1× TBS buffer pH 7 (50 mM Tris-HCl pH 7.4, 150 mM NaCl)
-
Salmon sperm
-
20× SSC
-
pH 7 (1× SSC = 3 M NaCl, 0.3 M sodium citrate pH 7)
-
Deionized formamide (Sigma F9037)
-
Tween 20 (Sigma P1379)
-
BSA (Sigma B8667)
-
Anti-digoxigenin antibody conjugated to fluorescein isothiocyanate (FITC) (Roche 11 207 741 910)
-
Extravidin conjugated to Cy3 (Sigma S6402)
-
4,6-Diamidino-2-phenylindole (DAPI; Roche 10 236 276 001)
-
Vectashield (Vector Laboratories)
Equipment
-
Vibratome series 1000plus (TAAB Laboratories Equipment Ltd)
-
Tube heater, PCR machine or water bath (for labeling probes)
-
Modified thermocycler (Omnislide; Hybaid Ltd) for pretreatment and ISH
-
Water bath (for posthybridization washing steps or for pretreatment instead of the modified thermocycler)
-
Humidity chamber set at 37 °C for probe detection (or ISH instead of the modified thermocycler) (see EQUIPMENT SETUP)
-
Confocal microscope equipped with suitable lasers (major providers: Leica Microsystems, Carl Zeiss, Nikon and Olympus Corporation) (see EQUIPMENT SETUP)
Reagent setup
-
Dextran sulfate Dissolve 5 g dextran sulfate ultrapure MW500000 in TE buffer to a final volume of 10 ml by stirring and heating at 70 °C. Aliquots of the solution can be stored at −20 °C for years.
-
NTB buffer For 500 μl stock: mix 250 μl 1 M Tris-HCl pH 7.5, 25 μl 1 M MgCl2, 125 μl 20 mg ml−1 BSA and 100 μl water. It can be stored at −20 °C for years.
-
Unlabeled dNTP mixture Mix 2 μl of each 100 mM dATP, dCTP and dGTP with 394 μl NTB buffer. It can be stored at −20 °C for months.
-
Dot blot buffer 1Mix 10 ml 1 M Tris-HCl pH 7.5, 5 ml 3 M NaCl and sterile water up to 100 ml. It can be stored at room temperature (20–22 °C) for months.
-
Dot blot buffer 2 0.5% (wt/vol) blocking reagent in buffer 1.
-
Dot blot buffer 3 Mix 10 ml 1 M Tris-HCl pH 9.5, 1 ml 1 M NaCl, 5 ml 1 M MgCl2 and sterile water up to 100 ml. It can be stored at room temperature for months.
-
Hybridization mix For 10 ml stock: 6.25 ml deoinized formamide, 2.5 ml 50% dextran sulfate, 1.25 ml 20× SSC and 0.15 ml 10% SDS. Mix compounds well. Aliquots of the solution can be stored at −20 °C for years.
-
Blocking solution Add 0.15 g BSA and 0.2% (vol/vol) Tween 20 to 3 ml 4× SSC.
-
Formaldehyde fixative Make an 8% (wt/vol) solution of paraformaldehyde in water. Stir the cloudy suspension on a hotplate at 60 °C and then add 1–2 drops of 1 M NaOH. The solution should immediately clear. Cool and dilute to 4% by adding an equal volume of 2× PEM buffer.
-
Detection buffer Mix 22.5 μl NTB and 17.5 μl BCIP. Add dot blot buffer 3 to give a total volume of 5 ml.
Critical
Make up immediately before use.
Equipment setup
-
Humidity chamber set at 37 °C for probe detection (or ISH instead of the modified thermocycler) A simple homemade humidity chamber can be made as follows: place filter papers saturated in water at the bottom of a plastic box. Put two glass rods on the top of the filter paper and place the slides on top of these; then put the lid on the box and place the box in an incubator at 37 °C. This forms a chamber with relative humidity of 65–75%.
-
Confocal microscope equipped with suitable lasers (major providers: Leica Microsystems, Carl Zeiss, Nikon and Olympus Corporation) For the best optical resolution, use an oil-immersion objective of high NA (1.30) with a ×63 or ×100 magnification. Good results can also be obtained with high NA (1.25) water-immersion objectives. For best optical results, the refractive index of the specimen, of the specimen-mounting medium and of the immersion medium at the objective should all be the same and should be correct for the objective; this is usually difficult to achieve.
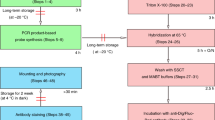
Procedure
Pretreatment of the slides
Timing ≈ 2 h
-
1
Wash the slides in 3% Decon for at least 1 h and rinse them thoroughly with distilled water. Air dry slides.
-
2
Coat the slides by dipping into a freshly prepared solution of 2% (vol/vol) APTES in acetone for 10 s.
-
3
Activate the slides by incubating with 2.5% (vol/vol) glutaraldehyde in phosphate buffer for 30 min, and rinse in distilled water and air dry slides.
Critical Step
Activate slides just before sectioning.
Fixation
Timing 1 h 30 min
-
4
Cut the chosen plant material and fix it in a 4% formaldehyde solution for 1 h at room temperature.
Critical Step
Vacuum infiltration is recommended to facilitate penetration of the fixative. Generally, good fixation is very important and a frequent source of problems. Immerse plant tissue in a plastic tube of fixative solution. The pressure above the solution should be reduced to approximately 20 kPa using a rotary vacuum pump, to facilitate uniform infiltration of the solution into the tissue. This minimizes the entrapment of air in the tissue that otherwise leads to incomplete infiltration. Bubbling of the solution around the tissue because of removal of the trapped gas is observed during vacuum infiltration, and completion of the infiltration is identified by termination of bubble formation in the fixative solution. This typically takes about 5 min per infiltration.
-
5
Wash samples in 1× TBS buffer for 10 min at room temperature.
Pause point
Fixed tissue can be stored at 4 °C for up to 4 d, but it is recommended to use it for sectioning within 1 d.
Sectioning
Timing Several days, depending on the number of samples
-
6
Cut 20-μm-thick sections from the plant tissue using a vibratome and allow to dry on multiwell slides.
-
7
Check the quality of the sectioned tissue by phase-contrast microscopy.
Critical Step
The quality of the sectioned tissue is crucial to obtain good hybridization signals. We recommend to check the quality of the sectioned tissue by phase-contrast microscopy. Section the plant tissue under sterile water using the vibratome by rapidly vibrating the knife blade. To get the best-quality sections, the amplitude of the blade vibration has to be maximized and the tissue has to be sectioned very slowly.
Pause point
Sectioned samples can be stored at 4 °C for several days. In some cases, they can be stored with desiccant at −20 °C for several weeks.
Probe labeling
Timing 2 h 30 min
-
8
Mix 5 μl 10× NTB buffer, 1 μl 100 mM DTT, 5 μl unlabeled dNTP stock, 1 μl dUTP-biotin (or dUTP-digoxigenin), 2–3 μg DNA template (total genomic DNA, repetitive sequences amplified by PCR or bacterial artificial chromosomes (BACs), 5 μl DNA polI/DNaseI, and sterile water up to 50 μl final volume.
Critical Step
The quality of probes is critical for ISH. It is strongly recommended to check the quality of the DNA in an electrophoresis gel before labeling it.
-
9
Incubate at 15 °C for 90 min in a water bath, tube heater or PCR machine.
-
10
Stop the labeling reaction by adding 5 μl 0.5 M EDTA pH 8 and cool on ice.
-
11
Purify the probe either using the MinElute PCR purification kit (A) or alternatively by precipitation overnight with 100% ethanol at −20 °C (B).
-
A
MinElute PCR purification kit
-
i
Add 5 volumes of buffer PB to 1 volume of the labeled probe and mix.
-
ii
Place a QIAquick spin column in the 2 ml collection tube provided.
-
iii
Apply the sample to the spin column and centrifuge at room temperature for 1 min at 17,900g.
-
iv
Discard the flow-through and place the spin column back in the same tube.
-
v
Wash the DNA by adding 0.75 ml of buffer PE to the spin column and centrifuge at room temperature for 1 min at 17,900g.
-
vi
Discard the flow-through, place the spin column back in the same tube and centrifuge the column at room temperature for an additional 1 min at 17,900g.
-
vii
To elute DNA, place the spin column in a clean 1.5 ml microcentrifuge tube, add 30 μl of sterile water, let the column stand for 1 min and then centrifuge at room temperature for 1 min at 17,900g.
-
i
-
B
Precipitation overnight with 100% ethanol at −20 °C
-
i
Add 1 ml of 100% ethanol to 1 volume of the labeled probe, mix vigorously and leave it overnight at −20 °C.
-
ii
Centrifuge the mix for 30 min at 4 °C at 12,000g.
-
iii
Discard the supernatant, wash the precipitate with 70% ethanol and centrifuge for 30 min at 12,000g.
-
iv
Discard the supernatant and leave the precipitate to dry at room temperature.
-
v
Dissolve DNA by adding 30 μl of sterile water.
Pause point
Labeled probes can be stored at −20 °C for months.
-
i
-
A
Checking the labeling reaction (dot blot test)
Timing 2 h
-
12
Cut a piece of Hybond N+ membrane and soak in dot blot buffer 1 for 5 min. Air dry the membrane.
-
13
Load the membrane with 1 μl of the DNA probe and leave to air dry for 5–10 min.
-
14
Place the membrane in dot blot buffer 1 for 1 min.
-
15
Place the membrane in dot blot buffer 2 for 30 min with shaking.
-
16
Incubate the membrane in (1:5,000, vol/vol in dot blot buffer 1) anti-digoxigenin-AP or (1:500, vol/vol in dot blot buffer 1) anti-biotin-AP solution for 30 min at 37 °C.
-
17
Wash the membrane in dot blot buffer 1 for 15 min.
-
18
Wash the membrane in dot blot buffer 3 for 2 min.
-
19
Incubate the membrane in the dark in detection buffer for 5 min (for color development).
-
20
Wash the membrane in water and leave to air dry.
FISH: prehybridization steps
Timing 2 h
-
21
Dehydrate slides in an ascending methanol/water series (30%, 50%, 70% and 100%) for 2 min each and air dry slides.
-
22
Incubate the tissue sections with the enzyme mix (1% (wt/vol) cellulase, 0.5% (wt/vol) pectolyase in TBS) for 1 h at 37 °C in humidity chamber.
-
23
Wash the tissue sections in TBS for 10 min.
-
24
Dehydrate slides in an ascending methanol/water series (30%, 50%, 70% and 100%) for 2 min each and air dry slides.
Critical Step
Do not store slides after the prehybridization treatment. Proceed with the ISH within hours.
FISH: hybridization
Timing overnight
-
25
Make 20 μl of hybridization mixture per well by adding 1 μl of (1 mg ml−1) salmon sperm and 1 μl of labeled probe to 18 μl of hybridization mix stock.
-
26
Denature the hybridization mixture at 95 °C for 5 min, cool on ice for another 5 min and immediately add 20 μl per well and put plastic coverslips over the wells.
Critical Step
Air-bubbles can hamper hybridization. To avoid these, apply the hybridization mixture by pipetting the hybridization mixture carefully in one step.
-
27
Denature slides and probe simultaneously in a modified thermocycler at 78 °C for 8 min.
Critical Step
Overdenatured preparations will result in poor chromosome morphology and/or loss of tissue. Underdenatured preparations might have very good chromosome morphology, but show weak or no signal.
-
28
Cool down slides progressively in the thermocycler (50 °C for 1 min, 45 °C for 1 min, 40 °C for 1 min and 38 °C for 5 min).
Critical Step
Cooling down probes and chromosomes gradually and simultaneously from the denaturation temperature to the hybridization temperature facilitates the annealing between the probe and the chromosomes, reduces background and also helps in renaturation of the chromosome structure.
-
29
Incubate slides in the thermocycler at 37 °C overnight. Alternatively, hybridization overnight can be done in the humidity chamber at 37 °C.
FISH: posthybridization steps and detection of the probes
Timing 3 h
-
30
Wash samples briefly at 42 °C in 2× SSC to allow coverslips to float off.
Critical Step
Do not allow slides to dry through Steps 30–39.
-
31
Wash slides in 20% (vol/vol) deionized formamide in 0.1× SSC for 10 min at 42 °C.
-
32
Wash slides in 2× SSC for 10 min at 42 °C.
-
33
Wash slides in 2× SSC for 10 min at room temperature.
-
34
Wash slides in 0.2% (vol/vol) Tween 20 in 4× SSC for 10 min at room temperature.
-
35
Incubate samples with the blocking solution for 5 min at room temperature.
-
36
Incubate samples with (1:100) anti-digoxigenin antibody conjugated to FITC or (1:500) extravidin-Cy3 (for digoxigenin-labeled probes or biotin-labeled probes, respectively) for 1 h at 37 °C in humidity chamber.
Critical Step
Air bubbles can hamper detection. Protect fluorochromes from the light, keeping them in the dark at 4 °C.
-
37
Wash slides in 0.2% (vol/vol) Tween 20 in 4× SSC for 15 min at room temperature.
-
38
Incubate samples with 20 μl of (1 μg ml−1) DAPI for 10 min at room temperature in the dark.
Critical Step
Air bubbles can hamper DAPI staining. Protect DAPI from light by keeping DAPI and stained slides in the dark at 4 °C.
-
39
Wash slides in 0.2% (vol/vol) Tween 20 in 4× SSC for 10 min at room temperature.
-
40
Mount slides in Vectashield or similar antifading mounting medium adding one small drop on each well and place a full-length glass coverslip over the slide, being careful to avoid air bubbles.
Critical Step
The use of antifading reagent is very important for imaging of fluorescent dyes. The choice of a specific antifading reagent depends on the label used. We found Vectashield to be the most suitable antifading reagent for FITC-labeled and Cy3-labeled DNA probes.
Pause point
Mounted slides can be stored in a dark and dry place at 4 °C for several days before image acquisition.
Image acquisition
Timing 1 h per sample
-
41
Collect confocal optical section stacks. Our most recent publications have used a Leica TCS SP confocal microscope equipped with a krypton and an argon laser, or have used a conventional wide-field microscope with a cooled CCD camera followed by image deconvolution. The topic of image acquisition is a large one, and the reader is referred to the extensive literature on this topic (e.g., refs. 17 and 18).
-
42
Analyze your data. In our work, we have transferred the recorded data to NIH image (a public domain program for the Macintosh by W. Rasband available via ftp from ftp://Zippy.nimh.nih.gov). Images have usually been composed for publication using Adobe Photoshop 5.0 (Adobe systems Inc.).
Troubleshooting
There are many variables in ISH protocols, making troubleshooting notoriously difficult. It is worth optimizing the whole protocol with a well-known probe to gain confidence in the procedure. We have summarized in Table 1 some of the most frequent reasons for poor results for an ISH in sectioned plant tissue.
Timing
Steps 1–3, pretreatment of the slides: estimated 2 h
Steps 4 and 5, fixation: 1 h 30 min
Steps 6 and 7, sectioning: several days, depending on the number of samples. Usually sectioning one sample takes between 30 min and 1 h
Steps 8–11, probe labeling: 2 h 30 min
Steps 12–20, checking the labeling reaction: 2 h
Steps 21–24, FISH: prehybridization steps: 2 h
Steps 25–29, FISH: hybridization: overnight
Steps 30–40, FISH: posthybridization steps and detection of the probes: 3 h
Steps 41–42, image acquisition: estimated 1 h per sample
Anticipated results
The use of the vibratome has been crucial for revealing detailed structures of internal plant cells that are not accessible to confocal microscopy on the intact plant or tissue. The thickness of sections that can be cut from unembedded plant tissue using a vibratome ranges from about 20 μm to 1 mm. Figure 1 (panel a) shows a low-magnification confocal image of a longitudinal section 1 mm thick along a wheat root showing the meristematic region. Apart from the caliptra and the apical meristem, all the internal tissues are visible and well preserved, even the central column of developing xylem vessel cells. Figure 1b shows a series of confocal images through a transverse section through a wheat floret, showing a single anther. Each anther contains four locules, each of which comprises the central pollen mother cells (PMCs) or microsporocytes and the surrounding tapetal cell layers. This specimen is at an early stage of meiosis.

All images are projections from confocal sections spaced 0.5 μm apart. (a) Low-magnification confocal image of a longitudinal rice root meristem vibratome section stained with DAPI. The caliptra and the apical meristem and all the internal tissues, including the central column of developing xylem vessel cells are visible (arrows). (b) Low-magnification confocal image of a wheat anther vibratome section stained with DAPI. A transverse section through the anther is shown, including four locules with the microsporocytes in the central area (arrows). (c) Telomere behavior during early meiosis in wheat. The projections have been produced to show the telomeres (red) in PMC at an early developmental stage. The telomeric probe was labeled with biotin and detected in red with anti-biotin-Cy3. In the PMC, telomeres are associated as a cluster on one side of the cell (arrows). (d) Homologous segment behavior at the telomere cluster stage during early meiosis in wheat. Rye segments were labeled with digoxigenin and detected with anti-digoxigenin FITC (green segments). (e) Overlay of c and d. (f) Telomere behavior during an earlier stage of meiosis in a wheat line carrying a small rye chromosomal introgression. Telomeres have just started to associate to form the telomere cluster. The two rye chromosome segments inserted into the wheat line are still decondensed and they can be seen as two foci. (g) Centromere behavior on diploid rice. The rice centromeric probe was labeled with digoxigenin and detected with anti-digoxigenin FITC. Centromeres are associated on one side of the nucleus in the developing xylem vessel cells (bigger cells, arrows) and they are dispersed on the nucleus in the rest of the root tissues. (h) Fluorescence in situ confocal images of a BAC for rice chromosome 1 in root sections of diploid rice. The BAC was labeled with biotin and detected with anti-biotin-Cy3 in red. (i) Telomere and heterochromatin behavior in an early meiotic nucleus in the diploid rye. Rye heterochromatin knobs are green and telomeres red. Scale bar, 100 μm (a), 10 μm (b–i).
Tissue sections for FISH are usually between 20 and 50 μm thickness. ISH can be used on thicker sections, but labeled probes do not penetrate efficiently to the innermost cells, and very thick specimens cause problems for high-resolution microscopy, even when a confocal microscope is used. FISH results, in common with all molecular cytogenetic techniques, strongly depend on the quality of the tissue that is used. Preservation and morphology of the intact nuclei are especially important to achieve good FISH results. Good penetration of the probes through the cell wall and into the nucleus is also very important when tissue sections are used for ISH experiments. Probe penetration is enhanced by dehydration in methanol and the treatment of the tissue sections with the cellulase and pectolyase enzyme mix, which partially digests cell walls. Probe penetration can be improved with a longer incubation with the enzyme mix, but a balance between digestion and structural preservation has to be found. Different plant species may require different cell wall digestion enzymes. Some examples of different ISH experiments are presented here to illustrate the range of possibilities this procedure has for investigation of plant nuclear organization, using the protocol described.
The onset of meiosis is marked by the development of a telomere cluster in the PMCs16. At this point in anther development, PMCs display only one or two nucleoli (DAPI-negative circular regions in the nucleus). Before the telomeres form a cluster, PMCs and tapetal cells are not morphologically distinguishable, and can be classified only by their position in the anther. To label telomeres, we have used the telomeric sequence as a probe, amplified previously by PCR using the oligomer primers (5′-TTTAGGG-3′)5 and (5′-CCCTAAA-3′)5 in the absence of template DNA19. Telomeres associated as a telomere cluster are shown in Figure 1c and e. Figure 1f shows a microsporocyte at a stage before the telomeres form a cluster. For the unequivocal identification and staging of the cells, the general morphology of the flower, the number of nucleoli present in the nucleus, the morphology of the nucleus, the position of the cell in the anther and the disposition of the telomeres can be used. This method has been used to analyze chromosome association during early meiosis in rice, wheat and related species2,3,5,15,16.
This procedure enables not only repetitive DNA structures such as centromeres, telomeres and heterochromatin knobs to be labeled, but also whole chromosomes or chromosome arms in nuclei containing DNA from more than one genome, as occurs in hybrids or in addition, translocation or substitution lines between related species. GISH has been previously used to assess chromosome association on intact nuclei3,16. We have used a wheat (T. aestivum cv. 'Chinese Spring') line carrying rye segments substituted for the equivalent region of the one-dimensional pair of wheat chromosomes. These chromosomes still have wheat telomeres and sub-telomeric regions, and the rye segment possesses a similar gene content but different repetitive content compared to the equivalent wheat region20. Vibratome anther sections were labeled simultaneously with total rye genomic DNA (green) and telomere probes (red) by ISH and visualized by 3D confocal microscopy (Fig. 1c–f). The two rye DNA segments are observed as elongated structures when telomeres are clustering (Fig. 1d,e). In the previous stage of meiosis, the two rye segments appear as two foci (Fig. 1f) as they do in somatic interphase nuclei.
Chromosome association has been investigated in cultivated rice (O. sativa, AA, 2n=24, cv. Bengal). ISH using the rice centromeric sequence (RCS2, GenBank accession no. AF058902) as probe was used to detect centromeres in the interphase nuclei of intact root sections in O. sativa. Results are shown in Figure 1g. Centromeres remain largely unassociated in this cell type.
BACs can also be detected by this protocol. A BAC from rice chromosome 1 was used as probe21 for ISH. The a0040G11 clone is located in contig 16 in rice chromosome 1 and mapped approximately at 100 cM21. ISH with this BAC in the root tissue of O. sativa clearly shows two hybridization sites for each BAC corresponding to the two homologous chromosomes (Fig. 1h). The sequence pSc250, amplified by PCR using total rye genomic DNA as a template to label rye heterochromatin knob DNA22, and the telomeric sequence were used for ISH on S. cereale cv. Petkus. Results were visualized by 3D confocal microscopy (Fig. 1i). Telomeres cluster on one side of the nucleus (red), whereas the heterochromatin knobs on each rye chromosome remain as tight foci (green).
Thus we have demonstrated that this procedure is suitable for analyzing a wide range of probes and tissues. The examples that we show in this article not only validate the procedure that we present, but also show that ISH carried out in 3D plant tissue is a powerful tool for investigating plant nuclear organization.
Accession codes
Change history
20 September 2007
In the version of this article initially published online, the e-mail address for Pilar Prieto appeared as 152prarm@uco.es; it should have been L52prarm@uco.es. The error has been corrected in the HTML and PDF versions of the article.
References
Aragon-Alcaide, L., Beven, A., Moore, G. & Shaw, P. The use of vibratome sections of cereal spikelets to study anther development and meiosis. Plant J. 7, 905–908 (1998).
Martinez-Perez, E., Shaw, P. & Moore, G. Polyploidy induces centromere association. J. Cell Biol. 148, 233–238 (2000).
Martinez-Perez, E., Shaw, P. & Moore, G. The Ph1 locus is needed to ensure specific somatic and meiotic centromere pairing. Nature 411, 204–208 (2001).
Prieto, P., Moore, G. & Reader, S. Control of conformation changes associated with homologue recognition during meiosis. Theor. Appl. Genet. 111, 505–510 (2005).
Prieto, P., Santos, A.P., Moore, G. & Shaw, P. Chromosomes associate premeiotically and in xylem vessel cells via their telomeres and centromeres in diploid rice (Oryzasativa). Chromosoma 112, 300–307 (2004).
Shaw, P.J., Beven, A.F., Leader, D.J. & Brown, J.W. Localization and processing from a polycistronic precursor of novel snoRNAs in maize. J. Cell Sci. 111, 2121–2128 (1998).
Rawlins, D.J., Highett, M.I. & Shaw, P.J. Localization of telomeres in plant interphase nuclei by in situ hybridization and 3D confocal microscopy. Chromosoma 100, 424–431 (1991).
Bunney, T.D. et al. Association of phosphatidylinositol 3-kinase with nuclear transcription sites in higher plants. Plant Cell 12, 1679–1688 (2000).
Wegel, E. et al. Three-dimensional modelling of wheat endosperm development. New Phytol. 168, 253–262 (2005).
Wegel, E., Vallejos, R.H., Christou, P., Stoger, E. & Shaw, P. Large-scale chromatin decondensation induced in a developmentally activated transgene locus. J. Cell Sci. 118, 1021–1031 (2005).
Thompson, W.F., Beven, A.F., Wells, B. & Shaw, P.J. Sites of rDNA transcription are widely dispersed through the nucleolus in Pisum sativum and can comprise single genes. Plant J. 12, 571–581 (1997).
Schwarzacher, T. et al. Genomic in situ hybridization to identify alien chromosomes and chromosome segments in wheat. Theor. Appl. Genet. 84, 778–786 (1992).
Prieto, P., Ramírez, M.C., Ballesteros, J. & Cabrera, A. Identification of intergenomic translocations involving wheat, Hordeum vulgare and Hordeum chilense chromosomes by FISH. Hereditas 135, 171–174 (2001).
Prieto, P., Martín, A. & Cabrera, A. Chromosomal distribution of telomeric and telomeric-associated sequences in Hordeum chilense by in situ hybridization. Hereditas 141, 122–127 (2004).
Prieto, P., Shaw, P. & Moore, G. Homologue recognition during meiosis is associated with a change in chromatin conformation. Nat. Cell Biol. 6, 906–908 (2004).
Martinez-Perez, E. et al. Homologous chromosome pairing in wheat. J. Cell Sci. 112, 1761–1769 (1999).
Pawley, J.B. (ed.) Handbook of Biological Confocal Microscopy 453–467 (Springer, New York, 1995).
Pawley, D. & Flinchbaugh, J. The current state: progress starts here. Manuf. Eng. 137, 71 (2006).
Cox, A.V. et al. Comparison of plant telomere locations using PCR-generated synthetic probe. Ann. Bot. 72, 239–247 (1993).
Moore, G. et al. Key features of cereal genome organization as revealed by the use of cytosine methylation-sensitive restriction endonucleases. Genomics 15, 472–482 (1993).
Chen, M.S. et al. An integrated physical and genetic map of the rice genome. Plant Cell 14, 537–545 (2002).
Vershinin, A., Schwarzacher, T. & Heslop-Harrison, J.S. The large-scale genomic organization of repetitive DNA families at the telomeres of rye chromosomes. Plant Cell 7, 1823–1833 (1995).
Acknowledgements
This work was funded by the Biotechnology and Biological Sciences Research Council of the UK. P.P acknowledges the support of Professor Antonio Martín.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Prieto, P., Moore, G. & Shaw, P. Fluorescence in situ hybridization on vibratome sections of plant tissues. Nat Protoc 2, 1831–1838 (2007). https://doi.org/10.1038/nprot.2007.265
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2007.265
This article is cited by
-
The nonpathogenic strain of Fusarium oxysporum FO12 induces Fe deficiency responses in cucumber (Cucumis sativus L.) plants
Planta (2023)
-
A rapid preparation procedure for laser microdissection-mediated harvest of plant tissues for gene expression analysis
Plant Methods (2019)
-
Chromatin-associated transcripts of tandemly repetitive DNA sequences revealed by RNA-FISH
Chromosome Research (2016)
-
Mapping the ‘breaker’ element of the gametocidal locus proximal to a block of sub-telomeric heterochromatin on the long arm of chromosome 4Ssh of Aegilops sharonensis
Theoretical and Applied Genetics (2015)
-
Simultaneous specific in planta visualization of root-colonizing fungi using fluorescence in situ hybridization (FISH)
Mycorrhiza (2014)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
