
Abstract
Thyrotropin-releasing hormone receptor type 2 (TRH-R2), not TRH-R1, has been proposed to mediate the CNS effects of TRH and its more effective analog taltirelin (TAL). Consistent with this idea, TAL exhibited higher binding affinity and signaling potency at mouse TRH-R2 than TRH-R1 in a model cell system. We used TRH-R1 knockout (R1ko), R2ko and R1/R2ko mice to determine which receptor mediates the CNS effects of TAL. There was no TRH-R1 mRNA in R1ko and R1/R2ko mice and no TRH-R2 mRNA in R2ko and R1/R2ko mice. Specific [3H]MeTRH binding to whole brain membranes was 5% of wild type (WT) for R1ko mice, 100% for R2ko mice and 0% for R1/R2ko mice, indicating TRH-R1 is the predominant receptor expressed in the brain. In arousal assays, TAL shortened sleep time with pentobarbital sedation in WT and R2ko mice by 44 and 49% and with ketamine/xylazine sedation by 66 and 55%, but had no effect in R1ko and R1/R2ko mice. In a tail flick assay of nociception, TAL increased response latency by 65 and 70% in WT and R2ko mice, but had no effect in R1ko and R1/R2ko mice. In a tail suspension test of depression-like behavior, TAL increased mobility time by 49 and 37% in WT and R2ko mice, but had no effect in R1ko and R1/R2ko mice. Thus, in contrast to the generally accepted view that the CNS effects of TAL are mediated by TRH-R2, these effects are mediated primarily if not exclusively by TRH-R1 in mice.
Similar content being viewed by others

INTRODUCTION
Administration of thyrotropin-releasing hormone (TRH) (pyroglutamyl-histidyl-prolinamide; pGlu-His-ProNH2) causes many CNS effects in rodents, including reversal of sedation induced by narcotics (‘arousal’), reduction in pain sensitivity (antinociception), and decreased depression-like behaviors (for review, see (Monga et al, 2008)). Many TRH analogs were synthesized that exhibited improved CNS activity (for review, see (Kinoshita et al, 1998)). TAL ((l-methyl-(S)-4,5-dihydroorotyl)-histidyl-prolinamide, TA-0910) is the only TRH analog approved for use in humans; it is used in Japan to treat patients with spinocerebellar degeneration (CeredistR). TAL exhibits higher stability in the blood and brain compared with TRH, and higher activity as an antagonist of pentobarbital-induced sedation than TRH in rodents. TAL exhibited modality-specific antinociceptive effects in mice also. Moreover, TAL displayed lower thyrotropin (TSH)-releasing activity in rodents than TRH. These findings were interpreted to suggest that TAL binds to different receptors in TSH-secreting cells of the pituitary than in the CNS.
There are two types of G protein-coupled receptors for TRH in rodents, subtype 1 (TRH-R1) and subtype 2 (Thyrotropin-releasing hormone receptor type 2, TRH-R2) (Sun et al, 2003). In humans, by contrast, only a single TRH receptor is expressed, which is more similar to TRH-R1 than TRH-R2 (Matre et al, 1993). It has been reported that TAL binds with lower affinity than TRH to receptors in rodent pituitary, most likely to TRH-R1, and in brain preparations, most likely to both TRH-R1 and TRH-R2 (Asai et al, 1999; Kinoshita et al, 1997). However, TAL pharmacology at each TRH receptor has not been characterized. Nevertheless, an hypothesis was developed that the CNS effects of TAL are mediated by TRH-R2 not TRH-R1 (for review, see Khomane et al, 2011). This hypothesis is based primarily on two observations. (1) TRH-R1 and TRH-R2 are expressed throughout the CNS, but in some nuclei only one receptor is expressed (O’Dowd et al, 2000). The brain areas mediating the CNS effects of TAL have been suggested to correlate with the unique sites of TRH-R2 expression. And (2), a number of TRH analogs have robust effects on the CNS, but minimal effects at the pituitary where TRH-R1 is the primary receptor (Gary et al, 2003).
In this study, we tested this hypothesis directly. We used TRH-R1 knockout (R1ko), R2ko, and R1/R2ko mice to determine, which receptor mediates the CNS effects of TAL. We found that the effects of TAL in arousal from narcotic sedation, in antinociception to thermal stimulus (tail flick assay), and in a behavioral assay for antidepressant activity (tail suspension test) were present in wild type (WT) and R2ko, but were absent in R1ko and R1/R2ko mice.
MATERIALS AND METHODS
Materials
Dulbecco’s modified Eagle’s Medium (DMEM) and fetal bovine serum were purchased from Biosource (Rockville, MD). TRH (pyroGlu-His-ProNH2) and MeTRH (pGlu-His(1(τ)-methyl)-ProNH2) were purchased from Sigma (St Louis, MO). [3H]MeTRH was purchased from PerkinElmer (Waltham, MA). TAL (N-[[(4S)-Hexahydro-1-methyl-2,6-dioxo-4-pyrimidinyl]carbonyl]-L-histidyl-L-prolinamide) was obtained from Tocris (San Diego, CA).
Cell Culture and Transfection
The generation of HEK-EM 293 (human embryonic kidney) cells stably expressing murine TRH-R1 or TRH-R2 was described earlier (Engel et al, 2008). HEK-EM cells stably expressing all TRH receptors were grown in DMEM containing 10% fetal bovine serum, 100 units/ml penicillin, 10 μg/ml streptomycin and 200 μg/ml hygromycin B (Invitrogen, Carlsbad, CA) at 37 °C in a humidified 5% CO2 incubator.
Preparation of Membranes from Cells and Mouse Brains
Whole brain tissues from mice were isolated and washed with 10 ml of phosphate-buffered saline (PBS). The tissues were homogenized in Tris-Mg buffer (TM buffer; 20 mM TrisCl and 2 mM MgCl2, pH 7.6) using a Dounce homogenizer. The nuclei and cell debris of homogenates were removed by centrifugation at 1000 g for 5 min at 4 °C. The postnuclear membrane fraction was collected from the supernatant by centrifugation at 40 000 g for 30 min at 4 °C and resuspended in TM buffer to store at −70 °C in aliquots.
Competition Binding
Competition binding assays were performed in monolayers of intact HEK cells expressing murine TRH-R1 (60 000 per cell) or murine TRH-R2 (180 000 per cell). The receptor number per cell was calculated from competition binding curves of various doses of unlabeled MeTRH and 4 nM [3H]MeTRH. The cells (220 000 cells per well in 24-well plates) were preincubated with various concentrations of unlabeled TAL, TRH or MeTRH for 15 min before addition of radioligand and then incubated at 37 °C for 1 h with 4 nM [3H]MeTRH as described elsewhere (Engel et al, 2006). Nonspecific binding was determined in incubations with excess nonradiolabeled MeTRH. IC50 is the concentration of unlabeled ligand that reduces specific binding of [3H]MeTRH by 50%.
To measure competition binding to membranes from the brain, a suspension containing 100 μg of membrane protein in TM buffer with 1% milk powder was incubated with 4 nM [3H]MeTRH and various concentrations of TAL, TRH, MeTRH, or cold MeTRH for 4 h on ice. The reaction mixture was diluted with the same ice cold buffer and passed through a Whatman GF/D filter. Filters were washed three times with ice cold TM buffer containing 1% milk powder, dried, and the retained radioactivity was measured.
Measurement of Intracellular Calcium Mobilization
Cells stably expressing a single TRH receptor were seeded in black-walled, clear-bottomed 96-well plates (Corning, NY) at a density of 60 000 cells per well in DMEM with 10% fetal bovine serum and incubated at 37 °C for 24 h in 5% CO2. The following day, the culture media was replaced with 100 μl of Hank’s Balanced Salt Solution with 20 mM HEPES, pH 7.5, and the cells were loaded with 100 μl of calcium 4 fluorescent dye (FLIPR Calcium 4 Assay Kit, Molecular Devices, Sunnyvale, CA) for 1 h at room temperature before the addition of compounds. Transient changes in intracellular [Ca++] induced by TAL, TRH, or MeTRH were measured using the FLIPRTETRA system (Molecular Devices, Sunnyvale, CA). Changes in fluorescence were detected at the emission wavelength ranges from 515 to 575 nm. The agonistic responses of ligands were assessed immediately upon their addition in a concentration range from 0.1 to 10 μM. Responses were measured as peak fluorescent intensity minus basal fluorescent intensity at each compound concentration.
Measurement of IP1 Production
Cells were seeded at 220 000/well in white, solid bottom, tissue culture-treated 24-well plates and cultured at 37 °C with 5% CO2 overnight. Serial dilutions of TAL, TRH or MeTRH in Hank’s Balanced Salt Solution with 20 mM HEPES and 50 mM LiCl, pH 7.4, were added at 200 μl/well on the second day. After incubation at 37 °C for 60 min in 5% CO2, IP1 content was measured using the IP-One ELISA kit (Cisbio International, France) according to the manufacturer’s protocol. The results were calculated as IP1 nmoles/well.
Mouse Studies
All experiments involving animals were approved by the Institutional animal care and use committee at the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda. TRH-R1+/− male mice in 129S1/SvImJ background were obtained (Omeros Corporation, Seattle, WA) and bred with WT 129S1/SvImJ females. Resulting heterozygous mice were bred to obtain homozygous TRH-R1−/− (R1ko) mice (Zeng et al, 2007). TRH-R2-deficient (R2ko) mice in a mixed 129/Svev background were generated and bred as reported (Sun et al, 2009). TRH-R1−/−/TRH-R2−/−-double-deficient (R1/R2ko) mice were generated by crossing R1ko with R2ko mice. Water and food (NIH-07; Zeigler Brothers) were available ad libitum. At the time of experiments, mouse ages ranged from 9 to 35 weeks, with a mean of 18.6±7.3 weeks, and mean body weight was 26.6±3.6 g. Subjects were tested only once per week, in a 7-week period. WT, R1ko, R2ko, and R1/R2ko mice all grew at similar rates and were fertile. The test regimen included arousal from pentobarbital sedation, arousal from ketamine-xylazine sedation, tail flick test and tail suspension test.
Arousal from Pentobarbital Sedation
To assess the analeptic effects of TAL, WT, R1ko, R2ko and R1R2ko mice (n>7/group; 20–30 g body weight) received intraperitoneal (ip) injections of either vehicle or TAL (1 mg/kg body weight), and 10 min later received pentobarbital sodium (50 mg/kg, ip). As each mouse lost its righting reflex, the animal was placed on its back and the time was measured until the righting reflex returned. When an animal was able to right itself so that it could consistently touch the cage with all four paws, three times in rapid succession, the righting reflex was considered regained. Sleeping time is defined as the duration of the righting reflex loss in minutes (Rinehart et al, 1986). Within 30 min after pentobarbital administration, some mice were found to be tremulous. As TRH has been found to induce tremors in WT mice (Ushijima et al, 1984), we filmed the mice during this time period so as to determine, which mice were tremulous after TRH receptor activation.
Arousal from Ketamine-Xylazine Sedation
WT, R1ko, R2ko, and R1R2ko mice were injected ip with ketamine (100 mg/kg)/xylazine (10 mg/kg) in PBS followed after 10 min with vehicle (PBS) or TAL (1 mg/kg body weight). The duration of sedation was the elapsed time after injection of vehicle or TAL until the mice regained the righting reflex as described above.
Tail Flick Assay
Antinociceptive effects of TAL were assessed using a radiant heat tail flick analgesia meter (Tail Flick Analgesia Meter Model 0104-301 M, Columbus Instruments, Columbus, OH) as described elsewhere (Bannon and Malmberg, 2007). Mice were placed in a restrainer 10 min after receiving vehicle or TAL (1 mg/kg, ip) and three tail flick measurements were made at 1–2 min intervals. Tails were positioned on the instrument, so that the radiant heat source was focused from 2 to 4 cm from the distal end of the tail. Maximal exposure time was set at 14 s to avoid tissue damage. The average of the three measurements was used as the tail-flick response latency for each mouse.
Tail Suspension Test
We followed modifications of the method by Steru et al, (Steru et al, 1985). Briefly, TAL (1 mg/kg) was administered ip 10 min before experimentation. Climbstoppers (4 cm long) were used to prevent tail climbing behavior (Can et al, 2012). Mice were individually suspended using adhesive tape by the distal quarter of their tails, in groups of four. The session was videotaped, starting before suspension and ending 6 min after last mouse was suspended. Mobility time over the first 6 min of suspension was scored for each mouse by at least three independent observers, trained in the method and blinded to the genotype.
Data Analysis
Data were analyzed by nonlinear regression curve fit using GraphPad Prism software version 4 (GraphPad, San Diego, CA) and the significance was determined by t-test or ANOVA.
RESULTS
We studied TAL binding and signaling in HEK-EM 293 cells engineered to express murine TRH-R1 or TRH-R2. We compared the effects of TAL and TRH in competition binding assays with [3H]MeTRH, a high affinity agonist for both TRH receptors (Sun et al, 2003), and on two early steps in signal transduction by TRH receptors (Gershengorn, 1986), inositol-1,4,5-trisphosphate production (by measuring its degradation product inositolmonophosphate) and intracellular calcium release (Figure 1 and Table 1). In competition binding assays with 4 nM [3H]MeTRH, the concentration of TRH or TAL that inhibited half maximal binding of [3H]MeTRH (IC50) to TRH-R1 was 520 nM for TAL and 15 nM for TRH. For TRH-R2, the IC50s were 110 nM for TAL and 5.4 nM for TRH. In experiments that measured stimulation of inositol-1,4,5-trisphosphate production and on intracellular calcium release, the half-maximally effective concentrations (EC50s) for inositolmonophosphate production were 72 nM for TAL and 4.0 nM for TRH at TRH-R1, and 2.7 nM for TAL and 0.52 nM for TRH at TRH-R2. For calcium release, the EC50s were 2.0 nM for TAL and 0.47 nM for TRH at TRH-R1, and 0.12 nM for TAL and 0.11 nM for TRH at TRH-R2. We showed that TAL is a moderately high affinity agonist at both receptors with selectivity for TRH-R2 over TRH-R1. The high potency of TAL relative to its apparent affinity is consistent with it being a ‘superagonist’ at mouse TRH receptors, as we have recently shown for TAL action at human TRH receptors (Thirunarayanan et al, 2012).

Binding and signaling of TAL to TRH-R1 and TRH-R2. Competition binding for [3H]MeTRH stimulation of inositol-1,4,5-trisphosphate production (by measuring its degradation product inositolmonophosphate) and stimulation of intracellular calcium release by various doses of TAL in HEK293 cells stably expressing TRH-R1 or TRH-R2 were measured as described in Materials and Methods. The maximal levels of IP1 in a typical experiment were: 2735±47 nmol for TRH at TRH-R1; 3590±91 nmol for TAL at TRH-R1; 3923±77 nmol for TRH at TRH-R2; and 3841±78 nmol for TAL at TRH-R2. The data represent the mean of 2–4 replicates in three experiments.
We used genetically modified mice to determine whether TRH-R1 or TRH-R2, or both, mediate the CNS effects of TAL. We studied previously reported TRH-R1 knockout (R1ko) (Zeng et al, 2007) and R2ko mice (Sun et al, 2009), and newly generated R1/R2ko mice and compared them to WT mice. TRH-R1 and TRH-R2 mRNA levels were similar in WT brains (Figure 2). As expected, there was no TRH-R1 mRNA detectable in R1ko and R1/R2ko mice and no TRH-R2 mRNA detectable in R2ko and R1/R2ko mice (Figure 2). We measured [3H]MeTRH binding to membranes isolated from whole brains of these mice (Figure 3). The level of specific [3H]MeTRH binding (total minus that in the presence of 30 μM MeTRH) was 5% of WT for R1ko mice, 100% for R2ko mice, and 0% for R1/R2ko mice, indicating that TRH-R1 is the predominant receptor expressed in the brain. TAL was able to compete as effectively as MeTRH for binding in all membrane preparations.

TRH-R1 and TRH-R2 mRNA levels. TRH-R1 and TRH-R2 mRNA was measured in the brains from two WT, R1ko, R2ko, and R1/R2ko mice as described in Materials and Methods. There was no detectable mRNA for TRH-R1 in R1ko and R1/R2ko mice, and for TRH-R2 in R2ko and R1/R2ko mice. Data are shown as mean±SD.

[3H]MeTRH binding to mouse brain membranes. Equal amounts of membrane protein were incubated at 4 °C with 2 nM [3H]MeTRH for 4 h without (Control) or with 30 μM MeTRH or 30 μM TAL. There was no difference in the binding in the presence of MeTRH or TAL. By t-test, specific binding (Control minus MeTRH) was found in membranes from WT and R2ko mice (***P<0.001) and in membranes from R1ko (++P<0.005), but not in membranes from R1/R2ko (P>0.1) mice. Data are shown as mean±SEM of five experiments.
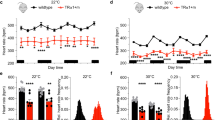
To test the hypothesis that TRH-R2 mediates the CNS effects of TAL directly, we measured the effect of TAL on CNS responses in four tests with all four mouse genotypes. The arousal response to TAL was determined in two tests by inducing sedation with pentobarbital in mice pretreated with TAL or PBS (control) or with ketamine/xylazine followed by the administration of TAL or PBS, and measuring the time of the loss of the righting reflex (‘sleep time’). TAL shortens sleep time in control animals (Suzuki et al, 1990). With pentobarbital alone, sleep times in control mice were similar in WT and R2ko mice but prolonged in R1ko and R1/R2ko mice, suggesting a role in arousal for endogenous TRH acting at TRH-R1. Sleep times with ketamine/xylazine alone were similar in all groups (Figure 4). Importantly, the results with TAL treatment did not comply with the prevailing hypothesis. TAL shortened sleep time in WT and R2ko mice by 44 and 49%, respectively, with pentobarbital sedation and by 66 and 55%, respectively, with ketamine/xylazine sedation. By contrast, TAL had no effect on sleep time in R1ko and R1/R2ko mice; there was a trend to a shorter sleep time in R1ko mice but it did not reach statistical significance.

Effects of TAL on arousal from pentobarbital (a) and ketamine/xylazine (b) sedation, latency in tail flick test (c) and mobility in tail suspension tests (d). The effects of TAL were tested in female and male mice separately. Because results were similar in females and males, the data were pooled. All graphs show mean±SEM. Control mice received vehicle (PBS) rather than TAL. (a) TAL shortened sleep time after pentobarbital sedation in WT (n=10) and R2ko mice (n=10) compared with their own Controls (***P<0.001), but not in R1ko (n=18) and R1/R2ko mice (n=10) (P>0.1). R1ko (n=10) and R1/R2ko mice (n=10) exhibited longer Control sleep times than WT (n=19) and R2ko mice (n=9) (+++P<0.001). (b) TAL shortened sleep time after ketamine/xylazine sedation in WT (n=10) and R2ko mice (n=9) compared with their own Controls (n=20, n=9) (***P<0.001), but not in R1ko (n=10) and R1/R2ko mice (n=10) compared with their Controls (n=8, n=10) (P>0.1). (c) TAL increased latency in the tail flick test in WT (n=10) and R2ko (n=9) mice compared with Controls (n=10) (**P<0.002), but not in R1ko (n=10) and R1/R2ko mice (n=10) (P>0.1). (d) TAL increased mobility time in the tail suspension test in WT (n=10) and R2ko mice (n=9) compared with Controls (n=10) (**P<0.002), but not in R1ko (n=10) and R1/R2ko mice (n=10) (P>0.1).
We measured the antinociceptive effects of TAL using a thermal stimulus (tail flick assay). The response latency is the time interval from administration of the stimulus to withdrawal of the tail from the stimulus. Latency time is increased by TAL in WT mice (Webster et al, 1983). TAL increased the response latency by 65 and 70%, respectively, in WT and R2ko mice but had no effect in R1ko and R1/R2ko mice (Figure 4).
The tail suspension test is a behavioral assay for assessing antidepressant activity in animals subjected to short-term inescapable stress that normally elicits a rapid adaptation to an immobile posture. Immobility time is reduced by the acute administration of antidepressants, and TAL was shown previously to produce a similar effect (Kinoshita et al, 1998). TAL increased mobility time (total minus immobility time) by 49 and 37%, respectively, in WT and R2ko mice but had no effect in R1ko and R1/R2ko mice (Figure 4).
To show that these effects are not specific to TAL, we measured the effects of TRH during pentobarbital-induced sedation. As TRH is not as active as TAL in causing arousal, we recorded inducement of tremors as a second measurement of the TRH effect (Ushijima et al, 1984). The arousal effect of TRH was less than that induced by TAL and the results were inconsistent. A more striking effect of TRH was its effect to induce tremors during pentobarbital-induced sedation. The effects of TRH and TAL to induce tremors in every WT and R2ko mice, but not in any R1ko and R1/R2ko mice are compiled in Table 2. Thus, it is likely that all TRH analogs primarily mediate their effects via TRH-R1.
DISCUSSION
In this study we directly tested the consensus view that the CNS effects of the TRH analog TAL are mediated by TRH-R2 in mice. Using genetically modified mice, we found that the arousal, tremor-inducing, antinociceptive, and antidepressant-like effects of TAL were lost in mice, in which TRH-R1 was deleted, R1ko and R1/R2ko mice, but were maintained in mice, in which TRH-R2 was deleted, R2ko mice. These data show conclusively that it is TRH-R1 and not TRH-R2 that primarily, if not exclusively, mediates these CNS actions of TAL. Moreover, we found that this is also the case for the effect of TRH itself to induce tremors during pentobarbital-induced sedation. These data, therefore, clearly contradict the idea that TRH-R2 is the predominant target that mediates TRH effects within the CNS.
Herein, we have studied the pharmacological effects of TAL and TRH. It was not a goal of this study to characterize the effects of deleting both TRH receptor subtypes in mice in the absence of TRH analog administration. Previous reports have described effects on CNS function in R1ko (Zeng et al, 2007) and R2ko mice (Sun et al, 2009). R1ko were found to exhibit increased depression-like and increased anxiety-like behaviors whereas female, but not male, R2/ko mice exhibited moderately increased depression-like and reduced anxiety-like phenotypes. We did observe that both R1ko and R1/R2ko, but not R2ko mice, display increased sleep time compared with WT mice when given pentobarbital to induce sedation. These findings suggest that TRH-R1 may be involved in maintaining a conscious state under normal physiological conditions. Also, R1ko mice were found to be mildly hypothyroid (Zeng et al, 2007), and this may have affected basal sleeping time in R1ko and R1/R2ko animals given pentobarbital. Of note for this study, however, is that the pharmacologic effects of TRH on pentobarbital-induced sedation are not affected by hypothyroidism (Breese et al, 1975). Further studies of the phenotype of mice with TRH receptor deletions are clearly indicated.
Although the data presented in this report demonstrate that at least some of the CNS effects of TAL and TRH are mediated by TRH-R1, there have been reports that effects of some TRH analogs may not be mediated by TRH-R1 or TRH-R2. Hinkle (Hinkle et al, 2002) studied the pharmacology of several TRH analogs, in which the His at position-2 was replaced by other amino acids, and found there was no correlation between the analeptic activities and the binding and signaling potencies of these analogs at TRH-R1 or TRH-R2. Hogan (Hogan et al, 2008) reported that a pentapeptide analog of TRH (pGlu-Asn-Pro-D-Tyr-D-TRPNH2) competed with [3H]MeTRH for binding to sites in the hippocampus and cortex, but did not bind to TRH-R1 or TRH-R2. The authors of both of these reports suggested that there may be sites other than TRH-R1 and TRH-R2 that serve as receptors for TRH analogs in the CNS. However, these other receptors, if they are present, have not been discovered.
As the human TRH receptor is homologous to TRH-R1, it is likely that the effects of TAL, TRH, and other TRH analogs in human subjects are mediated by the single TRH receptor. A long list of potential therapeutic uses for a TRHR agonist in humans has been reviewed (Yarbrough et al, 2007). However, reports of therapeutic benefits in many of these suggested uses, for example, effects as an antidepressant and in amyotrophic lateral sclerosis, have been accompanied by other studies, in which no therapeutic benefit has been found. It is possible that the inconsistent effects of TRH and its analogs in humans have been caused by rapid degradation of and poor blood-brain barrier penetration by these small peptides. If these pharmacokinetic characteristics were mitigating the potential therapeutic effects of these peptides, then small, lipophilic, drug-like agonists for TRHR may expose the therapeutic benefits of TRHR activation.
In conclusion, in contrast to the generally accepted view that the CNS effects of TAL and other TRH analogs are mediated by TRH-R2 or the less well accepted idea that these effects may be mediated by as yet undiscovered receptors in mice, we have shown that all of the CNS effects we studied are mediated primarily if not exclusively by TRH-R1. Future research will be directed at understanding the physiological roles of the TRH receptor subtypes.
References
Asai H, Kinoshita K, Yamamura M, Matsuoka Y (1999). Diversity of thyrotropin-releasing hormone receptors in the pituitary and discrete brain regions of rats. Jpn J Pharmacol 79: 313–317.
Bannon AW, Malmberg AB (2007). Models of Nociception: Hot-Plate, Tail-Flick, and Formalin Tests in Rodents. Curr Protoc Neurosci Chapter 8: Unit.
Breese GR, Cott JM, Cooper BR, Prange AJ, Lipton MA, Plotnikoff NP (1975). Effects of thyrotropin-releasing hormone (TRH) on the actions of pentobarbital and other centrally acting drugs. J Pharmacol Exp Ther 193: 11–22.
Can A, Dao DT, Terrillion CE, Piantadosi SC, Bhat S, Gould TD (2012). The tail suspension test. J Vis Exp 59: e3769.
Engel S, Neumann S, Kaur N, Monga V, Jain R, Northup J et al (2006). Low affinity analogs of thyrotropin-releasing hormone are super-agonists. J Biol Chem 281: 13103–13109.
Engel S, Skoumbourdis AP, Childress J, Neumann S, Deschamps JR, Thomas CJ et al (2008). A virtual screen for diverse ligands: discovery of selective G protein-coupled receptor antagonists. J Am Chem Soc 130: 5115–5123.
Gary KA, Sevarino KA, Yarbrough GG, Prange AJ, Winokur A (2003). The thyrotropin-releasing hormone (TRH) hypothesis of homeostatic regulation: Implications for TRH-based therapeutics. J Pharmacol Exp Ther 305: 410–416.
Gershengorn MC (1986). Mechanism of thyrotropin releasing hormone stimulation of pituitary hormone secretion. Annu Rev Physiol 48: 515–526.
Hinkle PM, Pekary AE, Senanayaki S, Sattin A (2002). Role of TRH receptors as possible mediators of analeptic actions of TRH-like peptides. Brain Res 935: 59–64.
Hogan N, O’Boyle KM, Hinkle PM, Kelly JA (2008). A novel TRH analog, Glp-Asn-Pro-D-Tyr-D-TrpNH2, binds to [3H][3-Me-His2]TRH-labelled sites in rat hippocampus and cortex but not pituitary or heterologous cells expressing TRHR1 or TRHR2. Neurosci Lett 431: 26–30.
Khomane KS, Meena CL, Jain R, Bansal AK (2011). Novel thyrotropin-releasing hormone analogs: a patent review. Expert Opin Ther Pat 21: 1673–1691.
Kinoshita K, Yamamura M, Sugihara J (1997). Distribution of thyrotropin-releasing hormone (TRH) receptors in the brain of the ataxic mutant mouse, rolling mouse Nagoya. Biol Pharm Bull 20: 86–87.
Kinoshita K., Yamamura M, Suzuki M, Matsuoka Y (1998). Taltirelin hydrate (TA-0910): an orally active thyrotropin-releasing hormone mimetic agent with multiple actions. CNS Drug Reviews 4: 25–41.
Matre V, Karlsen HE, Wright MS, Lundell I, Fjeldheim ÅK, Gabrielsen OS et al (1993). Molecular cloning of a functional human thyrotropin-releasing hormone receptor. Biochem Biophys Res Commun 195: 179–185.
Monga V, Meena CL, Kaur N, Jain R (2008). Chemistry and biology of thyrotropin-releasing hormone (TRH) and its analogs. Curr Med Chem 15: 2718–2733.
O’Dowd BF, Lee DK, Huang W, Nguyen T, Cheng R, Liu Y et al (2000). TRH-R2 exhibits similar binding but distinct regulation and anatomic distribution compared to TRH-R1. Mol Endocrinol 14: 183–193.
Rinehart RK, Barbaz B, Iyengar S, Ambrose F, Steel DJ, Neale RF et al (1986). Benzodiazepine interactions with central thyroid-releasing hormone binding sites: characterization and physiological significance. J Pharmacol Exp Ther 238: 178–185.
Steru L, Chermat R, Thierry B, Simon P (1985). The tail suspension test: a new method for screening antidepressants in mice. Psychopharmacology 85: 367–370.
Sun Y, Lu X, Gershengorn MC (2003). Thyrotropin-releasing hormone receptors - similarities and differences. J Mol Endocrinol 30: 87–97.
Sun Y, Zupan B, Raaka BM, Toth M, Gershengorn MC (2009). TRH-receptor-type-2-deficient mice are euthyroid and exhibit increased depression and reduced anxiety phenotypes. Neuropsychopharmacology 34: 1601–1608.
Suzuki M, Sugano H, Matsumoto K, Yamamura M, Ishida R (1990). Synthesis and central nervous system actions of thyrotropin- releasing hormone analogues containing a dihydroorotic acid moiety. J Med Chem 33: 2130–2137.
Thirunarayanan N, Raaka BM, Gershengorn MC (2012). Taltirelin is a superagonist at the human thyrotropin-releasing hormone receptor. Frontiers in Endocrinology 3: 1–4.
Ushijima I, Yamada K, Furukawa T (1984). Acute and long-term effects of thyrotropin releasing hormone on behavior mediated by dopaminergic and cholinergic activities in mice. Psychopharmacology 82: 301–305.
Webster VA, Griffiths EC, Slater P (1983). Antinociceptive effects of thyrotrophin-releasing hormone and its analogues in the rat periaqueductal grey region. Neurosci Lett 42: 67–70.
Yarbrough GG, Kamath J, Winokur A, Prange AJ (2007). Thyrotropin-releasing hormone (TRH) in the neuroaxis: therapeutic effects reflect physiological functions and molecular actions. Med Hypotheses 69: 1249–1256.
Zeng H, Schimpf BA, Rohde AD, Pavlova MN, Gragerov A, Bergmann JE (2007). Thyrotropin-releasing hormone receptor 1-deficient mice display increased depression and anxiety-like behavior. Mol Endocrinol 21: 2795–2804.
Acknowledgements
We thank Alexander Gregerov, PhD (Omeros Corporation, Seattle, WA) for generously providing the R1ko mice. This work was supported by the Intramural Research Program of the NIH, The National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), DK0011006.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Thirunarayanan, N., Nir, E., Raaka, B. et al. Thyrotropin-Releasing Hormone Receptor Type 1 (TRH-R1), not TRH-R2, Primarily Mediates Taltirelin Actions in the CNS of Mice. Neuropsychopharmacol 38, 950–956 (2013). https://doi.org/10.1038/npp.2012.256
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2012.256
