
Abstract
Toxic spills, hypoxia, disease outbreaks and toxin-producing algal blooms are all possible causes of mass mortality events, but in many cases it can be difficult to pinpoint the cause of death. Here we present a new approach that we name ‘forensic genomics’, combining field surveys, toxin testing and genomic scans. Forensic genomics queries allele frequencies of surviving animals for signatures of agents causing mass mortality and, where genetic diversity is high, is uniquely suited to identify natural selection in action. As a proof of concept, we use this approach to investigate the causes of an invertebrate mass mortality event, and its genetic effects on an abalone population. Our results support that a harmful algal bloom producing a yessotoxin was a major causative agent to the event.
Similar content being viewed by others

Introduction
Mass mortality events have been reported throughout the animal kingdom1,2,3,4,5, and in many cases it is difficult to pinpoint the agent responsible. One major cause of recent marine mortality events are harmful algal blooms (HABs)3,6,7,8 that kill either by toxin production or by hypoxia caused by decomposing algae8. Toxin-induced mortality is often difficult to identify since putatively causal toxins must be individually tested for levels and toxic effects in most cases9,10, as simultaneous screening of multiple toxins requires a significant infrastructure11. If the toxin is novel, toxicity data or presence/absence tests may be non-existent, making it difficult to infer mortality causation. However, it is possible that the surviving population may harbour a distinct genomic signature of toxin-induced natural selection, which would manifest as genetic variants (single nucleotide polymorphisms; SNPs) with abnormally high FST values in before–after comparisons12. In addition, gene functions related to the cause of mortality might be enriched in high FST SNPs. In many cases, there is some knowledge of the cellular targets of classes of potential toxins13,14, so by matching genomic data with what is known about putative causal toxins, it would in theory be possible to test hypotheses about the potential causes of a mortality event.
As a demonstration of this novel method, we examined a 2011 algal bloom-associated mass mortality event of a marine gastropod, the red abalone, Haliotis rufescens, in California15. This was a unique event, being the largest invertebrate die-off ever recorded in the region. Crabs, sea urchins and abalone were all affected simultaneously and showed high mortalities, whereas other taxa such as bat stars were not, suggesting an external mortality agent rather than a wholesale collapse of environmental quality such as hypoxia. Toxin testing was inconclusive, but suggested that a member of the yessotoxin (YTX) family could potentially be involved. YTX has a well-characterized mode of action affecting cytoskeleton organization (especially cadherin)14,16,17, the apoptosis pathway18,19,20, electron transport chain proteins19,21 and the immune system22. We hypothesized that YTX effects on abalone would manifest in SNP frequency changes at some of these pathways, whereas metabolic pathways of other classes of toxins would remain unchanged. For example, unlike common HAB toxins, such as microcystins or okadaic acid and its derivatives, YTXs do not inhibit protein phosphatases23, so if any of the above toxins were the cause of the mortality event, we would expect to see changes in protein phosphatases in before–after comparisons23,24. If hypoxia contributed to the mortality, we would also see changes in genes known to confer hypoxia tolerance (such as HIF-1a and arginine kinase)25,26 as well as enrichment for high FST SNPs in the gene ontogeny (GO) functional category ‘response to hypoxia’. In addition, as YTX is an organic cyclic compound24,27, we would expect the GO functional category ‘response to organic cyclic compound’ to be enriched for high FST SNPs if YTX was the cause. These hypotheses depend on the existence of standing genetic variation for response to YTX in abalone. However, the high levels of genetic polymorphism in abalone25 together with the potentially adaptive role of genetic diversity in loci involved in toxin tolerance in the natural environment suggest that this type of variation may exist. In addition, recent work showing that high-diversity invertebrates have genetic responses to other environmental stressors such as heat28 and acidification29 suggest that this might also be the case for toxin tolerance.
Although multiple causes cannot be ruled out, the genomic data support YTX being involved in the mass mortality event, through the effect of YTX on mitochondrial function and cadherin binding. Our data also implicate a well-known detoxification gene30, glutathione-S-transferase, in abalone toxin tolerance. Our results suggest that red abalones have genetic variation for response to YTX effects particularly in alleles at cadherin loci.
Results
Field surveys indicate high mortality
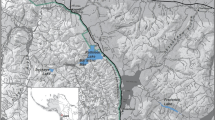
Field surveys at four different sites (Fig. 1) were conducted before and after the HAB event, which occurred at the end of August 2011. Mortalities ranged from 20 to 44.5% when averaged across three-depth zones (mean 25%), with the highest mortalities in shallow depth (<5 m, mean 48%, s.e. 13.5) (Fig. 2). Abalone densities dropped markedly at all four sites during the event and have remained low until present, prompting the California Fish and Game Commission to reduce the yearly take limit state-wide and to close the Fort Ross site completely in June 2013. Abalones in flow-through, oxygenated aquaria at a nearby marine laboratory were also killed, suggesting that hypoxia was not the cause of the event15. Known abalone diseases such as ‘Withering-foot syndrome31 were ruled out because dead animals lacked characteristic withering symptoms of the disease.

The geographical extent of the mortality event (marked with yellow lines) from Anchor Bay in the north to Bodega Bay in the south, as well as field survey locations indicated by red circles.

Mean transect percent mortality of red abalone along 30 × 2 m2 transects at the four survey sites in Sonoma County with s.d. error bars (abalone tissue samples for genomic work were taken from 0–5 m depth at Fort Ross).
Toxin testing inconclusive
Reports of an algal bloom in coastal waters during this time suggested that an algal-produced toxin might be responsible15. To test this, mussels, abalone and sea stars were tested for a wide range of known HAB toxins, including anatoxin-a, azaspiracids 1–5, domoic acid, lyngbyatoxin, microcystins, okadaic acid, saxitoxins, YTX and homo-YTX, with all but one showing no detectable levels (Supplementary Data 1). The exception was one class of algal toxins, YTX (maximum concentration 999 ng g−1), which was detected in the digestive glands of sickly abalone from Fort Ross. Additional testing found lower levels of YTX (15–559 ng g−1; n=15) in mussels and abalone digestive glands collected at Bodega Head, North Timber Cove, Potato Patch and Fort Ross. YTXs comprise a large family of toxins: at least 90 varieties of YTX have been isolated from a variety of phytoplankton and shellfish taxa32. Four variants are regulated in shellfish for human consumption by the European Union with a limit of 1,000 ng g−1 YTX27. However, nothing is known of YTX critical concentrations in invertebrates, and previous testing has shown YTX to be present up to 60 ng g−1 in non-bloom, non-mortality conditions33.
Forensic genomics implies Yessotoxin as cause of mortality
Previous to the die-off, we had surveyed the transcriptome of this abalone population for SNP markers25. The population has high genetic diversity and shows strong patterns of evolutionary response to local environmental conditions such as upwelling and acidification25. After the die-off, we were able to resample the population and compare transcriptomic-wide SNP frequencies pre- and post-mortality for signatures of natural selection at 15,016 loci. Using an FST outlier approach, we found 222 SNPs that were more divergent than expected under neutrality. Of these, 42 SNPs are within 34 annotated genes, which were more divergent than expected under neutrality (Fig. 3, Table 1, Supplementary Data 2), including genes related to cytoskeleton organization, electron transport chain, protein folding and response to organic cyclic compounds (Fig. 4, Supplementary Data 3) (high FST gene ontology over-representation analysis, Benjamini–Hochberg adjusted P-value <<0.001, nSNPs=27,610). Out of these, Se-cadherin (FST=0.215) shows a particularly strong series of changes. YTX is known to target cadherin14,17, causing a cascading effect of cytoskeleton disruption. In the Cadherin gene, the outlier SNP is located in the cytoplasmic region of the protein (pfam01049), responsible for catenin binding14. The outlier does not code for an amino-acid change. However, it is linked to a series of SNPs of unknown function in the 3′UTR, as well as to a series of silent SNPs up to 2,000 bp upstream that increased in frequency by 65% during the die-off (Fig. 5). The functional role of these changes is not known.

Plot of outlier SNP minor variant frequency versus FST, comparing mean outlier FST between randomly permuted populations from the original dataset (dotted line, nREPLICATES=100, error bars are 95% confidence intervals) and SNP outliers in the actual before–after comparison (dots and crosses; the crosses represent well-annotated SNPs, the remainder are marked as dots).

ReVIGO scatterplot of GO categories enriched for high FST SNPs (GO over-representation analysis (ORA), Benjamini–Hochberg corrected P<0.05, nSNPs=27,610), with darker circles denoting higher enrichment, circle size denoting breadth of the GO category and semantically similar (simRel) GO terms closer to each other (highly enriched (P<10−3) categories are marked in bold font).

Map of SNPs, genotypes and FST along the Cadherin contig (contig14782) in red abalone in Sonoma pre- and post-mortality, not including singletons and SNPs located in the UTR (amino-acid replacements are highlighted in blue).
Our data also show a multitude of cytoskeletal genes and functional categories related to cytoskeletal organization with outlier SNPs. In addition, cellular homeostasis and divalent metal-ion transport (P=0.03), as well as regulation of programmed cell death and apoptosis (P=0.03) are enriched for high FST SNPs. Another target seems to be mitochondrial function19,21, for which alleles at fructose-biphosphate aldolase (3 outliers with FST >0.1) seem to be especially related to toxin survivability. It is known that changes in intracellular calcium homeostasis disrupt mitochondrial membrane potential, inhibiting the electron transport chain24. Furthermore, glutathione-S-transferase, known in insects30 to be involved in detoxification of a wide range of exogenous compounds, contains three high FST SNPs, and may thus be a potential player in toxin detoxification in abalone.
We also examined alternative hypotheses regarding the metabolic pathways that may have been related to the die-off. The GO functional category ‘response to hypoxia’ was not enriched for high FST SNPs. Genes known to be involved in hypoxia tolerance (for example, HIF-1a (contig88699: nSNPs=27, Max FST=0.030), arginine kinase (contig90217: nSNPs=15, Max FST=0.033)) were not affected by the event as indicated by the FST outlier analysis. SNPs in protein phosphatase-coding sequences (N=67 across 17 sequences) were not affected by the mass mortality. Because there were no detectable levels of other toxins, such as azaspiracids, saxitoxin, domoic acid, okadaic acid, microcystins, lyngbyatoxin and anatoxin-a, these were effectively ruled out as the drivers of the mortality event.
Discussion
The methods used in this study, combining field studies, toxin testing and genetic forensics, can be used not only to determine the cause of death but also to enhance our understanding of the mechanisms of action at the organismal level. In this case, we can show that the population genomic effects of the mass mortality event are consistent with YTX being a major causative agent, while the combination of toxin testing and genetic sampling could rule out most other HAB toxins as well as hypoxia as main causes (although other factors could have certainly contributed to the event). This method currently depends on the existence of transcriptome-ready samples before a mortality event, a requirement which may not be met in most cases. However, as DNA-based methods of whole-genome analysis at the population level become more affordable, it may be possible to base before–after comparisons on DNA samples from museums or from population genetic monitoring programs.
Out of 222 detected FST outliers, only 42 could be reliably annotated, leaving 180 SNPs with unknown function being affected by the event. These might be contaminants or artifacts of an imperfect assembly, but they might also be of biological importance. As a consequence, we cannot completely rule out additional stressors as contributing to the event. However, as almost all annotated outliers are consistent with YTX as a cause, we can still conclude that the major causative agent was the toxin.
It is still unclear why mortalities were greater in shallow waters. It could potentially be due to higher toxin concentrations near the coast34, or from wave action increasing the solubility of the toxin. The reduction in mortality at depth is consistent with YTX as a causative agent, as the toxin may have been more concentrated (that is, at toxic concentrations) in shallow depths due to lower water volume and reduced mixing. However, it is important to note that very little is known about the mechanism of action of YTXs, making it difficult to draw specific conclusions about this. Another group of toxins that have similar effects to YTXs is azaspiracids. However, none of the organisms known to produce these toxins (Azadinium spp.) were present in the water at the time, and analysis of tissue samples for azaspiracids 1–5 was negative. The presence of two varieties of YTX, confirmed through testing, makes this the prime candidate of the mortality event.
In theory35,36,37, a reduction in genetic diversity and an increase in linkage disequilibrium can be seen around selected regions. Unfortunately, we do not know enough about linked regions in the abalone transcriptome. Within single contigs, it is possible to study linkage effects; within the Se-cadherin contig, for example, there are distinct blocks of linked loci visible (Fig. 5). However, without prior knowledge of linkage in the red abalone population, it is hard to draw general conclusions about the selective effects on linkage properties. The outlier analysis suggests a few loci with relatively high FST values increasing during the event from very low allele frequencies, estimated at 0 (denoted with asterisks in Table 1). Although it might be possible that these were present in low frequencies before the event and were selected for, uncertainty in the allele frequency estimation might also account for this pattern. Our sample size of 21–23 per population, while suggested to be adequate for decent allele frequency estimation38, might still be too low for low-frequency alleles. Therefore, we have chosen not to focus on these outliers.
By gaining insight into the ultimate targets of toxins, and the allelic basis of resistance to them, it may be possible to better identify more resistant natural populations. Such resilience mapping has recently been initiated for coral heat resistance28 and sea urchin acidification resistance29. One management strategy is to protect resilient populations from other sources of mortality, such as habitat destruction or fishing mortality for which they have no resistance. These protected populations may then be able to provide sustained population growth under future climate change scenarios, or contribute to a more productive aquaculture brood stock. Finally, we would urge any future comprehensive ecosystem monitoring programme to include routine acquisition of genetic libraries. The enhanced genetic knowledge gained before a mortality event occurs provides the genetic baseline, which can be compared with the genetics of the population post mortality thereby aiding in identification of the causative agent, as has been done in this example. We suggest that obtaining genetic baseline data is an important part of baseline monitoring of healthy wild populations.
Methods
Mortality field surveys
In September and October 2011 after the mass mortality of marine invertebrates was observed in Sonoma County, a series of three subtidal SCUBA surveys were conducted. Surveys were based at important red abalone fishing grounds within Sonoma County from south to north at Fort Ross (38° 30.1′ N, 123° 13.8′ W), Timber Cove (38° 31.8′ N, 123° 16.4′ W), Ocean Cove (38° 33.1′N, 123° 18.4′ W) and Salt Point (38° 33.7′ N, 123° 19.5′ W) (Fig. 1). Surveys were conducted by experienced subtidal research teams that counted live and dead marine invertebrates along 30 × 2 m2 transects at three-depth strata (0–5, 5.1–10 and 10.1–20 m) per site. Dead invertebrates were clearly differentiated from live as abalone being unattached to the substrate, upside down with an opaque white foot muscle tissue. Dead sea urchins were not attached to the substrate with spines fallen off the test and dead sea stars had curled-up arms with a white dead colouration to the flesh. The percent of dead invertebrates at each depth and site were recorded.
Toxin testing
A total of 40 tissue samples were collected between 27 August and 14 September 2011 from the Fort Ross area, Salt Point, Bodega Head, Bodega Harbor, North Timber Cove and Potato Patch (Supplementary Data 1). Samples were shipped frozen to UC Santa Cruz and were kept at −80 °C until processing. Approximately 1 g for each of 32 tissue samples was homogenized with 100% methanol (MeOH), which was subsequently diluted to 50% before cleanup. Samples were then split for toxin analysis of domoic acid, microcystins LR, RR, YR and LA, okadaic acid, saxitoxins, lyngbyatoxin, YTX and homo-YTX. Methanol extracts stored (−20 °C) were also subsequently tested for azaspiracids and re-tested for YTXs. Methodological details for toxins other than YTXs are not reported in detail since samples were negative for all other toxins; these analyses followed standard protocols39,40 using LC–MS for all but saxitoxins, which were screened using commercial enzyme-linked immunosorbent assay kits.
Samples for YTX and homo-YTX were initially screened by following the method of Paz et al. 41 with the following modifications. Homogenate (5 ml, 0.5 g tissue) was loaded onto BakerBond C18 cartridges and eluted in 90% MeOH. Samples were analysed on an Agilent 6130 liquid chromatography–mass spectrometry (LC–MS) system with an Agilent Poroshell 120 SB-C18 2.7-μm (2.1 × 50 mm2) column with electro-spray ionization in negative mode41. Selected ion monitoring of parent and daughter ions for YTX and homo-YTX was quantified with concentrations determined by comparison with standard curves for YTX and homo-YTX using certified standards (NRC, Canada).
From the initial screening, 12 samples were selected for further analysis representing potentially positive and negative samples from mussels, abalone (gill, digestive gland/gonad, foot) and sea star (gonad tissue from the arm). The LC–MS method was adapted to the protocol by These et al.42 and the tissue samples were hydrolysed42. This provided better separation and a lower limit of detection than the initial screening, with a limit of detection for total toxins of 20 ng g−1 tissue (calculated as 3.3 × s.d./S, where s.d. is the standard deviation and S is the slope of the calibration curve). Archived extracts were subsequently analysed using the same method, but without the hydrolyzation step, for all samples. The method detection limit for these samples was slightly lower than for the hydrolysed samples, at 15 ng g−1 tissue. Hydrolysed samples (n=4) were of 8–83% higher concentration than non-hydrolysed samples. For reporting purposes, homo-YTX and YTX were summed as total YTX. We did not screen for 45-hydroxy-YTX or 45-hydroxy-homo-YTX (the other two compounds regulated by the European Union). These have toxic equivalence factors of 1.0 and 0.5, so our results are potentially conservative estimates of total toxicity.
DNA sequencing and bioinformatics
Mantle tissue samples were collected from 24 live red abalone before the bloom (22 April 2011) and 24 abalone after the bloom (17 October 2011) at shallow depths in Fort Ross State Park, Sonoma County, California (38° 30.1′ N, 123° 13.8′ W) (Fig. 1). Total RNA was extracted and cDNA libraries were created from poly-A-selected mRNA. All samples were tagged with barcoded adaptor sequences and sequenced, six individuals per lane in an Illumina HiSeq 2000, for a total of eight lanes. One sample from before the event and one from after the event produced low-read counts, leaving 23 from before and 23 from after for further analysis. Using the protocol of De Wit et al.43, reads were filtered from low-quality base calls and residual adaptor sequences, and were then aligned using Burrows-Wheeler Aligner (BWA)44 to a recently published red abalone mantle transcriptome25. SNP detection using GATK45 resulted in 1,434,378 high-quality (P>0.99) SNP calls, out of which 46,063 had high-quality genotypes (P>0.99) for all individuals. A principal components analysis (Fig. 6a) showed that two individuals from before the event were significantly (10−65) different from all others in Principal Component (PC) one. Further investigation found that these individuals (FR 39, FR49) were heterozygous at all of the top 500 scoring loci in the PCA, raising the possibility of them being hybrids between red abalone and another co-occurring abalone species. These individuals were removed from the dataset and the SNP detection was rerun, resulting in 1,405,746 high-quality SNPs, out of which 45,966 had high-quality genotypes at all loci. A principal components analysis (Fig. 6b) indicated that there was no overall divergence between the before and after populations.

(a) Principal components analysis (PCA) of red abalone individuals before and after the die-off, indicating that individuals FR39 and FR49 were genetically distinct from remaining individuals; they were thus omitted from further analyses. (b) PCA of the same data, after removing individuals FR39 and FR49.
An FST outlier analysis was conducted using Lositan Selection Workbench46 after removing SNP loci obviously out of Hardy–Weinberg equilibrium and loci with low minor allele frequencies (<5%) (n=15,016). SNPs located above the 95% confidence interval of the overall FST distribution were considered as outliers, after which a false discovery rate correction of 0.05 was applied (Supplementary Data 2). To examine the outlier FST distribution compared with what would be expected from stochastic processes, we permuted the original dataset into 100 random replicates of two populations with 21 individuals each and plotted the outlier FST distribution (means and 95% confidence intervals) against heterozygosity for all permuted population pairs. The outliers are in all but one case (FST=0.05) highly significantly above the random outlier FST distribution.
All of the 34 annotated genes could be assigned open-reading frames using the OrfPredictor online software47 (available at http://proteomics.ysu.edu/tools/OrfPredictor.html), using the output of a BLASTx to NCBI’s nr database as supporting data. Based on these, all outlier SNPs were identified as synonymous, non-synonymous or untranslated (Supplementary Data 2).
An over-representation analysis was performed using ErmineJ48 based on the gene ontology annotation of the transcriptome, using an FST of 0.08 as a cut-off (based on the FST outlier permutation data). Functional categories significantly enriched in high FST SNPs were graphed using the online GO visualization software ReVIGO49.
Additional information
How to cite this article: De Wit, P. et al. Forensic genomics as a novel tool for identifying the causes of mass mortality events. Nat. Commun. 5:3652 doi: 10.1038/ncomms4652 (2014).
Accession codes: DNA sequences have been deposited in the NCBI short read sequence archive (SRA) under the accession code SRP037568. A text file listing all identified polymorphisms and individual genotypes at all loci is available on DRYAD (http://datadryad.org), doi:10.5061/dryad.2qs35.
Accession codes
References
Suttle, C. A. Marine viruses—major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812 (2007).
Peterson, C. H. et al. Long-term ecosystem response to the Exxon Valdez oil spill. Science 302, 2082–2086 (2003).
Scholin, C. A. et al. Mortality of sea lions along the central California coast linked to a toxic diatom bloom. Nature 403, 80–84 (2000).
Harvell, C. D. et al. Ecology—Climate warming and disease risks for terrestrial and marine biota. Science 296, 2158–2162 (2002).
Garrabou, J. et al. Mass mortality in Northwestern Mediterranean rocky benthic communities: effects of the 2003 heat wave. Glob. Change Biol. 15, 1090–1103 (2009).
Flewelling, L. J. et al. Red tides and marine mammal mortalities. Nature 435, 755–756 (2005).
Miller, M. A. et al. Evidence for a novel marine harmful algal bloom: Cyanotoxin (microcystin) transfer from land to sea otters. PLoS ONE 5, e12576 (2010).
Diaz, R. J. & Rosenberg, R. Spreading dead zones and consequences for marine ecosystems. Science 321, 926–929 (2008).
Rossini, G. P. Functional assays in marine biotoxin detection. Toxicology 207, 451–462 (2005).
Van Dolah, F. M. Marine algal toxins: Origins, health effects, and their increased occurrence. Environ. Health Perspect. 108, 133–141 (2000).
These, A., Klemm, C., Nausch, I. & Uhlig, S. Results of a European interlaboratory method validation study for the quantitative determination of lipophilic marine biotoxins in raw and cooked shellfish based on high-performance liquid chromatography-tandem mass spectrometry. Part I: collaborative study. Anal. Bioanal. Chem. 399, 1245–1256 (2011).
Narum, S. R. & Hess, J. E. Comparison of FST outlier tests for SNP loci under selection. Mol. Ecol. Resou. 11, 184–194 (2011).
Franchini, A., Malagoli, D. & Ottaviani, E. Targets and Effects of Yessotoxin, Okadaic Acid and Palytoxin: A Differential Review. Mar. Drugs 8, 658–677 (2010).
Ronzitti, G., Callegari, F., Malaguti, C. & Rossini, G. P. Selective disruption of the E-cadherin-catenin system by an algal toxin. Br. J. Cancer 90, 1100–1107 (2004).
Rogers-Bennett, L. et al. Dinoflagellate bloom coincides with marine invertebrate mortalities in northern California. Harmful Algae News 46, 10–11 (2012).
Korsnes, M. S., Hetland, D. L., Espenes, A. & Aune, T. Cleavage of tensin during cytoskeleton disruption in YTX-induced apoptosis. Toxicol. In Vitro 21, 9–15 (2007).
Callegari, F. & Rossini, G. P. Yessotoxin inhibits the complete degradation of E-cadherin. Toxicology 244, 133–144 (2008).
Malaguti, C., Ciminiello, P., Fattorusso, E. & Rossini, G. P. Caspase activation and death induced by yessotoxin in HeLa cells. Toxicol. In Vitro 16, 357–363 (2002).
Korsnes, M. S., Hetland, D. L., Espenes, A. & Aune, T. Induction of apoptosis by YTX in myoblast cell lines via mitochondrial signalling transduction pathway. Toxicol. In Vitro 20, 1419–1426 (2006).
Korsnes, M. S. & Espenes, A. Yessotoxin as an apoptotic inducer. Toxicon 57, 947–958 (2011).
Bianchi, C. et al. Yessotoxin, a shellfish biotoxin, is a potent inducer of the permeability transition in isolated mitochondria and intact cells. Biochim. Biophys. Acta 1656, 139–147 (2004).
Malagoli, D., Casarini, L. & Ottaviani, E. Algal toxin yessotoxin signalling pathways involve immunocyte mussel calcium channels. Cell. Biol. Int. 30, 721–726 (2006).
Ogino, H., Kumagai, M. & Yasumoto, T. Toxicologic evaluation of Yessotoxin. Nat. Toxins 5, 255–259 (1997).
Tubaro, A., Dell'Ovo, V., Sosa, S. & Florio, C. Yessotoxins: A toxicological overview. Toxicon 56, 163–172 (2010).
De Wit, P. & Palumbi, S. R. Transcriptome-wide polymorphisms of Red Abalone (Haliotis rufescens) reveal patterns of gene flow and local adaptation. Mol. Ecol. 22, 2884–2897 (2012).
Kawabe, S. & Yokoyama, Y. Role of Hypoxia-Inducible Factor α in response to hypoxia and heat shock in the Pacific oyster Crassostrea gigas. Mar. Biotechnol. 14, 106–119 (2012).
Paz, B. et al. Yessotoxins, a group of marine polyether toxins: An overview. Mar. Drugs 6, 73–102 (2008).
Barshis, D. J. et al. Genomic basis for coral resilience to climate change. Proc. Natl Acad. Sci. USA 110, 1387–1392 (2013).
Pespeni, M. H. et al. Evolutionary change during experimental ocean acidification. Proc. Natl Acad. Sci. USA 110, 6937–6942 (2013).
Shi, H. X. et al. Glutathione S-transferase (GST) genes in the red flour beetle, Tribolium castaneum, and comparative analysis with five additional insects. Genomics 100, 327–335 (2012).
Chambers, M. D., VanBlaricom, G. R., Hauser, L., Utter, F. & Friedman, C. S. Genetic structure of black abalone (Haliotis cracherodii) populations in the California islands and central California coast: Impacts of larval dispersal and decimation from withering syndrome. J. Exp. Mar. Bio. Ecol. 331, 173–185 (2006).
Miles, C. O. et al. Evidence of numerous analogs of yessotoxin in Protoceratium reticulatum. Harmful Algae 4, 1075–1091 (2005).
Howard, M. D. A., Silver, M. & Kudela, R. M. Yessotoxin detected in mussel (Mytilus californicus) and phytoplankton samples from the US west coast. Harmful Algae 7, 646–652 (2008).
McPhee-Shaw, E. E., Nielsen, K. J., Largier, J. L. & Menge, B. A. Nearshore chlorophyll-a events and wave-driven transport. Geophys. Res. Lett. 38, (2011).
Jensen, J. D., Thornton, K. R., Bustamante, C. D. & Aquadro, C. F. On the utility of linkage disequilibrium as a statistic for identifying targets of positive selection in nonequilibrium populations. Genetics 176, 2371–2379 (2007).
Kim, Y. & Nielsen, R. Linkage disequilibrium as a signature of selective sweeps. Genetics 167, 1513–1524 (2004).
Utsunomiya, Y. T. et al. Detecting Loci under Recent Positive Selection in Dairy and Beef Cattle by Combining Different Genome-Wide Scan Methods. PLoS ONE 8, e64280 (2013).
Hale, M. L., Burg, T. M. & Steeves, T. E. Sampling for Microsatellite-Based Population Genetic Studies: 25 to 30 Individuals per Population Is Enough to Accurately Estimate Allele Frequencies. PLoS ONE 7, e45170 (2012).
Lehane, M., Braña-Magdalena, A., Moroney, C., Furey, A. & James, K. Liquid chromatography with electrospray ion trap mass spectrometry for the determination of five azaspiracids in shellfish. J. Chromatogr. A 950, 139–147 (2002).
Miller, M. A. et al. Evidence for a novel marine harmful algal bloom: Cyanotoxin (microcystin) transfer from land to sea otters. PLoS ONE 5, e12576 (2010).
Paz, B., Riobo, P., Ramilo, I. & Franco, J. M. Yessotoxins profile in strains of Prorocentrum reticulatum from Spain and USA. Toxicon 50, 1–17 (2007).
These, A., Scholz, J. & Preiss-Wiegert, A. Sensitive method for the determination of lipophilic marine biotoxins in extracts of mussels and processed shellfish by high-performance liquid chromatography-tandem mass spectrometry based on enrichment by solid-phase extraction. J. Chromatogr. A 1216, 4529–4538 (2009).
De Wit, P. et al. The simple fool's guide to population genomics via RNA-Seq: an introduction to high-throughput sequencing data analysis. Mol. Ecol. Resour. 12, 1058–1067 (2012).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 25, 1754–1760 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Antao, T., Lopes, A., Lopes, R. J., Beja-Pereira, A. & Luikart, G. LOSITAN: A workbench to detect molecular adaptation based on a Fst-outlier method. BMC Bioinformatics 9, 323 (2008).
Min, X. J., Butler, G., Storms, R. & Tsang, A. OrfPredictor: predicting protein-coding regions in EST-derived sequences. Nucleic Acids Res. 33, (suppl 2): W677–W680 (2005).
Lee, H. K., Braynen, W., Keshav, K. & Pavlidis, P. ErmineJ: tool for functional analysis of gene expression data sets. BMC Bioinformatics 6, 269 (2005).
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 6, e21800 (2011).
Acknowledgements
We thank the California Department of Fish and Game for providing funding for sequencing, the NOAA ECOHAB Rapid Response program and the Central and Northern California Ocean Observing System for funds to complete toxin testing and PISCO for funding for laboratory materials. For help with sample collection, we also thank Christy Juhasz and members of the recreational abalone fishing community.
Author information
Authors and Affiliations
Contributions
P.D.W. is responsible for the study design, data analysis and manuscript writing. L.R.-B. was in charge of field surveys and funding for sequencing, R.M.K. performed all the toxicological work, whereas S.R.P. was responsible for funding for lab work and project management.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Data 1
Samples screened for algal toxins, and the subset of samples quantified for YTX and homo-YTX. Sample indicates the CDFW sample code; Toxins Screened include Anatoxin-a (A), Domoic Acid (D), Microcystins LR, RR, YR, LA (M), Lyngbyatoxin (L), Saxitoxin (S), Yessotoxin (Y), and Azaspiracids 1-5 (Z). (XLSX 48 kb)
Supplementary Data 2
Outlier SNPs listed by LOSITAN, open reading frames (ORFs) and amino acid changes. ORFs were assigned using OrfPredictor (http://proteomics.ysu.edu/tools/OrfPredictor.html) supplemented with data from a BLASTx against NCBI's nr database (e<10-5). (XLSX 29 kb)
Supplementary Data 3
GO categories significantly (p<0.05) enriched for high Fst SNPs after multiple test corrections. (XLSX 13 kb)
Rights and permissions
About this article

Cite this article
De Wit, P., Rogers-Bennett, L., Kudela, R. et al. Forensic genomics as a novel tool for identifying the causes of mass mortality events. Nat Commun 5, 3652 (2014). https://doi.org/10.1038/ncomms4652
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms4652
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
