
Abstract
The Flaviviridae are a family of viruses that cause severe human diseases. For example, dengue virus (DENV) is a rapidly emerging pathogen causing an estimated 100 million symptomatic infections annually worldwide1. No approved antivirals are available to date, and clinical trials with a tetravalent dengue vaccine showed disappointingly low protection rates2. Hepatitis C virus (HCV) also remains a major medical problem, with 160 million chronically infected patients worldwide and only expensive treatments available3. Despite distinct differences in their pathogenesis and modes of transmission, the two viruses share common replication strategies4. A detailed understanding of the host functions that determine viral infection is lacking. Here we use a pooled CRISPR genetic screening strategy5,6 to comprehensively dissect host factors required for these two highly important Flaviviridae members. For DENV, we identified endoplasmic-reticulum (ER)-associated multi-protein complexes involved in signal sequence recognition, N-linked glycosylation and ER-associated degradation. DENV replication was nearly completely abrogated in cells deficient in the oligosaccharyltransferase (OST) complex. Mechanistic studies pinpointed viral RNA replication and not entry or translation as the crucial step requiring the OST complex. Moreover, we show that viral non-structural proteins bind to the OST complex. The identified ER-associated protein complexes were also important for infection by other mosquito-borne flaviviruses including Zika virus, an emerging pathogen causing severe birth defects7. By contrast, the most significant genes identified in the HCV screen were distinct and included viral receptors, RNA-binding proteins and enzymes involved in metabolism. We found an unexpected link between intracellular flavin adenine dinucleotide (FAD) levels and HCV replication. This study shows notable divergence in host-dependency factors between DENV and HCV, and illuminates new host targets for antiviral therapy.
Similar content being viewed by others


Main
CRISPR is revolutionizing the use of genetic screens because the ability to completely knockout genes substantially increases the robustness of the phenotypes5,6. We compared the CRISPR approach in the hepatocyte cell line Huh7.5.1 with an alternative method to generate knockout alleles on a genome-wide scale: insertional mutagenesis in human haploid cells (HAP1)8,9 (Fig. 1a). Both methods generate libraries of cells with knockout mutations in all non-essential genes. To comprehensively identify cellular genes with crucial roles in the Flaviviridae life cycles, we first infected pools of mutagenized cells with DENV serotype 2 (DENV-2). The two types of genetic screening methods showed a strong concordance in the genes enriched in the DENV-2-resistant population. Many could be functionally classified into three distinct categories, each important for proper expression of ER-targeted glycoproteins (Fig. 1b, c, Supplementary Tables 1, 2). The translocon associated protein (TRAP) complex (containing subunits SSR1, SSR2 and SSR3) has an elusive role in stimulating co-translational translocation mediated by several, but not all, signal sequences10 (Fig. 1b, c, blue). Genes involved in protein quality control and the ER-associated protein degradation (ERAD) pathway also scored highly (Fig. 1b, c, green). Notably, in both the haploid and CRISPR genetic screens, the most significantly enriched genes were subunits of the OST complex, an enzyme essential for N-linked glycosylation (Fig. 1b, c, red). This dependence on ER cellular genes is probably related to the expression of the DENV genome, which encodes an ER-targeted viral polyprotein containing signal sequences and viral glycoproteins. Given the similarities in DENV and HCV polyprotein expression, we expected these genes to also be represented in the HCV CRISPR screen. Surprisingly, there was no overlap between the DENV and HCV core sets of enriched genes, suggesting that these members of the Flaviviridae evolved divergent host factor dependencies (Fig. 1c–e, Extended Data Fig. 1a, b, Supplementary Tables 3, 4). Indeed, cross-comparison of the most significant hits with both viruses suggested specific dependencies, although minor quantitative effects cannot be excluded (Extended Data Fig. 1c). The robustness of the CRISPR approach was further underscored by the consistent identification of the core dependency factors in three independent replicate screens performed for each virus (Extended Data Fig. 2). We validated the novel DENV host factors in isogenic knockout cells using a plaque-forming assay and observed a marked reduction in particle formation (Extended Data Figs 3, 4a). Importantly, complementation of knockout cells restored DENV infection (Extended Data Fig. 4b, c). The relevance of the identified host factors was further confirmed in Raji DC-SIGN, a B-cell line commonly used to study DENV (Extended Data Fig. 4d).

a, Schematic for genome-wide screening approach. NGS, next-generation sequencing. b, Haploid genetic screen for DENV host factors. The y axis represents significance of enrichment of gene-trap insertions in genes in DENV-resistant population compared to unselected HAP1 cells. Each circle represents a specific gene and size corresponds to the number of independent gene-trap insertions. All genes with P < 0.05 (Fisher’s exact test) were coloured and grouped by function. The screen was performed once. c, d, CRISPR genetic screen for DENV (c) and HCV (d) host factors in Huh7.5.1 cells. Significance of enrichment was calculated by RIGER analysis. The screens were performed in three replicates and the mean of the RIGER score is represented on the y axis. The 30 most enriched genes were coloured and grouped by function. e, Comparison of the 30 most enriched genes from the DENV and HCV CRISPR screens and their position based on the mean RIGER score.
Struck by the distinct host factor requirements of DENV-2 and HCV, we sought to evaluate selected DENV-2-dependency factors against other mosquito-borne flaviviruses that are closely related to DENV (Fig. 2a). Using quantitative PCR (qPCR) in isogenic knockout cells, we found that West Nile virus (WNV), but not yellow fever virus (YFV) or Zika virus (ZIKV), was as sensitive as DENV-2 to the disruption of the tested ERAD genes, which is in line with previous reports implicating ERAD in WNV infection11,12. A functional TRAP complex is important for DENV-2, YFV and ZIKV RNA replication, whereas WNV RNA abundance is only slightly reduced. Individual subunits of the OST complex displayed notably different phenotypes for the four related flaviviruses. Whereas knockout of STT3A and STT3B both completely abolished DENV-2 replication, only STT3A knockout affected YFV, WNV and ZIKV replication. When probing HCV replication in STT3A- and STT3B-knockout Huh7 cells using luciferase virus, we did not observe a substantial decrease (Extended Data Fig. 4e).

a, qPCR of DENV (clone 16681), YFV (17D), WNV (Kunjin), and ZIKV (Uganda) RNA in knockout HAP1 cells. WT, wild type. b, qPCR of prototypic strains of DENV serotypes 1–4 RNA in knockout (KO) Huh7 cells. c, Confocal microscopy of STT3B-KO Huh7 cells immunostained for DENV envelope (DENV-E) protein immediately or 30 min after DENV infection. Original magnification, ×630. d, Luminescence of DENV replicon RNA expressing luciferase in knockout Huh7 cells. The DENV NS5GDD mutant served as replication-deficient control. RLU, relative light units. Data are mean and s.e.m. (qPCR) or s.d. (RLU) for triplicate infections.
Intrigued by the differential sensitivity to the catalytic OST subunits, we focused our mechanistic studies on the OST complex, which has not been linked to viral replication before. The highly conserved catalytic subunit of the OST complex, STT3, is duplicated into two paralogues STT3A and STT3B in mammalian cells, and each isoform is present in distinct protein complexes13 (Extended Data Fig. 4f). The STT3A complex is important for the co-translational N-linked glycosylation of most of the glycoproteins, whereas the STT3B complex is important for the co-translational or post-translational glycosylation of acceptor sites that have been skipped by the STT3A complex14. Despite their partially redundant functions in N-linked glycosylation, we found that all DENV serotypes required the presence of both catalytic subunits as well as MAGT1, the highest scoring gene in the CRISPR screen (Fig. 2b).
To pinpoint which step in the viral life cycle requires the OST complex, we first focused on viral entry. We did not observe major differences in viral particle entry in OST-deficient cells (Fig. 2c). Next, we used a replicon assay that bypasses viral entry by electroporation of DENV RNA. Translation of the viral genome, apparent at time points up to 10 h, was equally efficient in OST-knockout cells as in wild-type cells (Fig. 2d). In stark contrast, viral RNA replication (apparent at time points after 10 h) was completely abolished. This mirrored the expression pattern observed with a replication-deficient dengue mutant in the viral polymerase (NS5GDD). Thus, we show that the OST complex has a crucial involvement in viral RNA replication, after entry and translation of the viral genome.
Most glycoproteins can be efficiently modified by both OST isoforms and only a few are preferentially modified by either STT3A or STT3B14. Concordant with this, knockout of either STT3A or STT3B did not lead to loss of cellular viability, whereas a double knockout was lethal (Fig. 3a). We demonstrated that OST catalytic activity is required for cellular function using STT3A and STT3B mutants containing mutations in the residues that coordinate the binding of the divalent cation required for catalysis15 (Fig. 3a, Extended Data Fig. 5). The functional redundancy between isoforms in global N-linked glycosylation is in contrast with the extreme dependence on each individual isoform of the OST complex for DENV replication. To investigate whether the effect of the OST complex is mediated by the necessity to glycosylate viral proteins properly, we focused on NS1, an enigmatic DENV glycoprotein with essential roles in RNA replication16 and pathogenesis17. NS1 was fully glycosylated in STT3A- and STT3B-knockout cells in contrast to the hypo-glycosylation of control cellular proteins known to be preferentially glycosylated by STT3A (pSAP) or STT3B (SHBG) (Fig. 3b). This led us to speculate that the OST complex itself rather than its catalytic activity is required for DENV replication. To explore this hypothesis, we used the catalytic mutants of STT3A and STT3B (Fig. 3a, c, Extended Data Fig. 5). Surprisingly, the catalytically dead mutants were able to restore fully DENV replication in STT3A- and in STT3B-knockout cells (Fig. 3d). We thus conclude that DENV RNA replication has hijacked a function of the human OST complex that is independent from its canonical role in N-linked glycosylation. The dispensability of the catalytic function of the OST complex further suggests a more structural role of OST in viral replication. The OST forms a stoichiometric complex at the ER membrane where DENV establishes a functional replication complex to initiate RNA replication. Our electron microscopy studies using APEX2 confirmed the localization of STT3B in the ER membrane, in close proximity to the membranous replication vesicles in DENV-2-infected cells (Extended Data Fig. 6a–c).

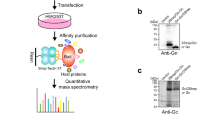
a, Viability of STT3A and STT3B double-knockout cells complemented with wild-type (WT) or catalytic (cat) mutant cDNA. Dox, doxycycline. b, Glycosylation of DENV protein NS1, SHBG and pSAP in STT3A- and STT3B-knockout Huh7 cells. Different glycoforms are indicated by arrowheads. Tun, tunicamycin. c, Glycosylation state of pSAP and SHBG in STT3A- and STT3B-knockout cells complemented with catalytic mutants. d, DENV infection of knockout Huh7 cells complemented with wild-type or catalytic mutants of STT3A and STT3B. Data are mean and s.d. of triplicate infections. e, Co-immunoprecipitations of STT3A–Flag and STT3B–Flag from DENV-infected cell lysates. LE, long exposure.
We next interrogated the physical interaction of the viral proteins with the OST complex by immunoprecipitation of STT3A–Flag and STT3B–Flag in the context of viral infection. Western blot analysis after immunoprecipitation showed that the non-structural proteins NS2B and NS3, components of the DENV replication complex, associate specifically with STT3A and STT3B (Fig. 3e, Extended Data Fig. 6d). Next, eluates of the immunoprecipitations (Extended Data Fig. 6e, f) were subjected to tryptic proteolysis and the resulting peptides were analysed by mass spectrometry. Identification of tryptic peptides from NS2A, NS3 and NS4A in the eluates pointed to their association with the OST complexes formed by STT3A and STT3B (Extended Data Fig. 6g, Supplementary Table 5). Taken together, this data suggests a structural role for the OST complex in DENV RNA replication through interactions with non-structural proteins that form the RNA replication complex. Our data indicate that the OST complex fulfils specialized roles in host pathogen interactions and is more multifaceted than previously recognized. In support of this emerging notion, two recent studies uncovered unexpected roles of the OST complex in immunity. The OST complex was found to be crucial for innate immune responses triggered by lipopolysaccharide18, and in a separate study OST dysregulation was identified as cause for autoimmune disorders triggered by certain TREX1 mutations19.
The HCV-resistant cell population was highly enriched in guide RNAs targeting the known HCV receptors20 CD81, OCLN and CLDN1, confirming their non-redundant role in entry for HCV (Fig. 1d, blue) and highlighting the validity of the screen results. The completeness of the screen was further underscored by the identification of microRNA-122 (ref. 21) and DGCR8 (Fig. 1d, green), which is part of the microRNA processing machinery, as key factors for HCV replication. Several dependency factors of HCV were validated in Huh7.5.1 cells, where knockout significantly reduced viral RNA and particle formation (Fig. 4a, b, Extended Data Figs 7, 8a). After CLDN1, the second most significantly enriched gene was ELAVL1 (also known as HUR), an RNA-binding protein involved in mRNA stabilization22. In isogenic ELAVL1-knockout cells, HCV RNA replication was nearly abolished, while we did not observe a decrease with other RNA viruses (Extended Data Fig. 8b, c) including the alphavirus Sindbis, which contains strong ELAVL1-binding sites23. We used an HCV replicon assay to show that ELAVL1 has a critical role in HCV RNA replication, which is in line with a recent report24 (Extended Data Fig. 8d, e). Enzymatically active HCV-dependency factors (Fig. 1d, red) are by far the most important category of host factors for identifying antiviral drug targets. A case in point is cyclophilin A (PPIA) that has been actively pursued until phase III clinical trials25. We discovered enzymes with novel putative roles in HCV replication and explored these potentially ‘druggable’ factors further. We focused on RFK and FLAD1, enzymes involved in the two-step conversion of riboflavin (vitamin B2) to flavin adenine dinucleotide (FAD) (Fig. 4c). RFK- and FLAD1-knockout cells were resistant to HCV replication but not DENV (Extended Data Fig. 9a). As predicted from their sequential role in FAD biogenesis, exogenous flavin mononucleotide (FMN) or FAD rescued HCV replication in RFK-knockout cells, whereas FAD but not FMN rescued viral replication in FLAD1-knockout cells (Fig. 4d, Extended Data Fig. 9b). This demonstrates that HCV replication is dependent solely on sufficient FAD levels. Modulation of intracellular FAD levels can be achieved by treatment of the cells with lumiflavin, a cellular uptake inhibitor of riboflavin26. Treatment of cells with lumiflavin greatly reduced HCV replication, while other RNA viruses were less sensitive to lumiflavin treatment (Fig. 4e, Extended Data Fig. 9b–f). We further pinpointed RNA replication as the step of the life cycle that requires FAD using a replicon system (Fig. 4f). This highlights that knockout screens can identify specific host targets for antiviral drug discovery. Taken together, we used comparative genome-scale knockout screens to identify human genes with crucial roles in the replication of Flaviviridae. Despite previous extensive interrogation of human host factors for these viruses through genomic and proteomic approaches11,27,28,29,30, we discovered marked dependencies on several host processes that had not been linked to flaviviral replication before (Fig. 4g, Extended Data Fig. 10).

a, qPCR of HCV RNA in Huh7.5.1 cell lines. b, HCV particle formation measured by focus-forming units (FFU) assay. ND, no foci detected (threshold of detection is 50 FFU ml−1). c, Biosynthesis pathway of FAD. Lumiflavin (LF) competitively inhibits uptake of riboflavin. Vit.B2, vitamin B2. d, qPCR of HCV RNA in untreated, FMN- or FAD-treated RFK- and FLAD1-knockout Huh7.5.1 cells. e, qPCR of DENV or HCV RNA in lumiflavin-treated Huh7.5.1 cells. For each concentration, the significance of the effect on HCV versus DENV was determined. f, HCV replicon assay in untreated and lumiflavin-treated Huh7.5.1 cells using wild-type sgJFH1 replicon. g, Model of identified DENV and HCV host factors. Data are mean and s.e.m. (qPCR) or s.d. (FFU, RLU) for triplicate infections. *P < 0.05, **P < 0.01, ***P < 0.001 (unpaired, parametric two-sided Student’s t-test, with Welch post-correction). NS, non-significant.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Haploid genetic screen
The haploid genetic screen was performed as previously described9. In short, 100 million gene-trap mutagenized HAP1 cells were seeded and infected with DENV-2 16681 (multiplicity of infection (MOI) = 5). Eight hours after infection, media was aspirated and replaced with IMDM containing 2% FBS. Clear cytopathic effects were observed after 3 days of infection leading to the death of most cells. Clusters of cells resistant to DENV-2 infection became apparent during further culture, and 9 days after infection cells were collected as a pool (yield ~30 million cells) and genomic DNA was isolated using a QIAamp DNA column. Gene-trap insertion sites were determined by linear amplification of the genomic DNA (gDNA) flanking regions of the gene-trap DNA insertion sites and sequenced on a Genome Analyzer II. Reads were aligned to the human genome using Bowtie and enrichment of independent insertions was calculated as previously described9.
CRISPR genetic screens
Huh7.5.1 cells were stably transduced with lentiCas9-Blast and subsequently selected using blasticidin. Next, a total of 300 million Huh7.5.1 cells that constitutively express Cas9 were transduced with the lentiGuide-Puro from the GeCKO v2 library31 at a MOI of 0.3. Cells were selected with puromycin and pooled together. The CRISPR genetic screens were started 10 days after transduction. Approximately 60 million mutagenized cells for each library (A and B) were infected with DENV-2 16681 (replicate 1 and 2) or DENV-2 strain 429557 (replicate 3) using a MOI of 1, or with HCV JFH1 at a MOI of 0.3. Cytopathic effect was visible 2 and 5 days after infection for DENV and HCV, respectively. Huh7.5.1 cells grow slower than HAP1 cells and clusters of resistant cells took longer to develop. The selected cells were collected 16 days after infection. As an uninfected reference we chose the unselected starting population because in these strong positive selection screens the selection pressure of the viral infections renders potential small growth differences of the mutagenized cells inconsequential. For both selected and uninfected control cells, gDNA was isolated using a QIAamp DNA column, and the inserted guide RNA sequences were amplified from the gDNA by flanking primers and prepared for next-generation sequencing. Resulting amplicons were sequenced on a MiSeq or NextSeq platform (Illumina) and the enrichment of each guide RNA was calculated by comparing the relative abundance in the selected and unselected population. RIGER analysis was performed on guide RNAs (with at least 10 reads) ranked by enrichment using the weighted sum statistical method32. Each CRISPR screen was performed in three replicates and the mean of the three RIGER scores was calculated.
Cell culture
HAP1 cells were derived from the near-haploid chronic myeloid leukaemia cell line KBM7 as described earlier9. HAP1 cells and knockout derivatives were cultured in IMDM supplemented with 10% FBS, penicillin-streptomycin and l-glutamine. STT3A/STT3B double-knockout HAP1 cells were cultured in IMDM supplemented with 10% FBS, penicillin-streptomycin, l-glutamine and 25 ng ml−1 doxycycline. Huh7, Huh7.5.1 (both gifts from F. Chisari) and HEK293FT (Thermo Scientific) cells and knockout derivatives were grown in DMEM supplemented with 10% FBS, penicillin-streptomycin, non-essential amino acids and l-glutamine. HEK293FT cells were used to generate lentivirus vectors for cellular transductions. Raji DC-SIGN cells (a gift from E. Harris) were cultured in RPMI-1640 supplemented with 10% FBS, penicillin-streptomycin and l-glutamine. The cell lines have not been authenticated. Parental cell lines have been tested negative for mycoplasma.
Viral serotypes and strains
DENV-2 infectious clone 16681 was a gift from K. Kirkegaard. DENV-2 from infectious clone 16681 was adapted to HAP1 cells through serial passaging. Viral whole-genome sequence analysis revealed three coding mutations compared to the original clone 16681: Q399H in envelope, L180F in NS2A and S238F in NS4B. DENV-1 Hawaii 1944 (#NR82), DENV-2 strain 429557 (#NR-12216), DENV-2 New Guinea C 1944 (#NR-84), DENV-3 Philippines/H871856 (#NR-80) and DENV-4 H241 Philippines 1956 (#NR-86) were ordered from BEI resources (NIH, NIAID). Yellow fever virus was generated by culturing the yellow fever YF-VAX 17D-204 vaccine. West Nile virus (Kunjin strain CH 16532) was a gift from J. F. Anderson. Zika virus (strain MR766) was provided by S. Weaver and R. Tesh. Hepatitis C virus JFH1 and HCV-Luc pFL-J6/JFH-5′C19Rluc2AUbi vector were gifts from C. Rice. Sindbis virus (SINV) strain Ar-339 (TC adapted) Egypt 1952 (ATCC VR-1585) and human rhinovirus 14 (ATCC VR-284) were ordered from the American Type Culture Collection. Poliovirus type 1 strain Mahoney was a gift from H. Ploegh. Venezuelan equine encephalitis virus TC-83 (pVEEV/GFP) was a gift from I. Frolov.
qPCR infectivity assays
Cells were plated in 96-well plates and infected with an MOI of 0.1 of virus, unless otherwise stated. Cells were collected as outlined in Ambion Power SYBR Green Cells-to-Ct kit (Ambion 4402954). Cells were collected 8 h after infection with polio virus, 24 h after infection with Sindbis virus, Venezuelan equine encephalitis virus and human rhinovirus 14, 2 days after infection with DENV-2 16681 and Zika virus, yellow fever virus, and West Nile virus Kunjin strain, 3 days after infection with HCV JFH1 and 5 days after infection with DENV-2 New Guinea. For comparison of DENV serotypes, cells were infected at an MOI of 0.01 with DENV-1 Hawaii 1944, DENV-2 New Guinea C 1944, DENV-3 Philippines/H871856 and DENV-4 H241 Philippines 1956 for 2 days. All samples were normalized to 18S expression. Two independent experiments were performed with triplicate infections and one representative is shown.
The following qPCR primers were used: DENV-2-forward: 5′-GCCCTTCTGTTCACACCAT-3′, reverse: 5′-GGCTCTGCCAATCAGTTCAT-3′; universal-DENV-forward: 5′-GGTTAGAGGAGACCCCTCCC-3′, reverse: 5′-GGTCTCCTCTAACCTCTAGTCC-3′; yellow fever forward: 5′-GAAATGCCTGCCCTTTATGA-3′, reverse: 5′-GCACATGGCAACAGAAGCTA-3′; Kunjin-forward: 5′-GCTTTGCCACCTCTCTTCAC-3′, reverse: 5′-CGGTTGATGGTTTCCACTCT-3′; ZIKV-forward: 5′-ACCATACGGCCAACAAAGAG-3′, reverse: 5′-TCCACAGCCAGGAAGAGACT-3′; HCV-forward: 5′-TCTCTCAGTCCTTCCTCGGA-3′, reverse: 5′-AAGCCGGCTAGAGTCTTGTT-3′; SINV-forward: 5′-CGCGGTCACGTAAGGATAAT-3′, reverse: 5′-TTTGGCATTCTTCAGCACAG-3′; polio-forward: 5′-CAACCTCCCACTGGTGACTT-3′, reverse: 5′-ATTTCCCCTGCTCAACCTTT-3′; 18S-forward: 5′-AGAAACGGCTACCACATCCA-3′, reverse: 5′-CACCAGACTTGCCCTCCA-3′; VEEV-forward: 5′-CAGGACGATCTCATTCTCAC-3′, reverse: 5′-TCATTCACCTTGTACCGAACG-3′; HRV-14-forward: 5′-AAGCAATTTGGTGGTCCAAG-3′, reverse: 5′-ACACTGGGGTTTGAAGCACT-3′.
Crystal violet staining
For virus infections, wild-type and knockout cell lines were plated out in either 24- or 96-well plates. Cells were infected with DENV-2 (16681), HCV, SINV or polio using a MOI of 1. Huh7-STT3A-KO-STT3B-KO-pLenti-TRE3G-CMV-STT3B cells were cultured in presence or absence of 25 ng ml−1 doxycycline. Cells were incubated for 48–120 h then fixed using 4% formaldehyde in PBS. Cell viability at time of fixation was determined by crystal violet staining.
Plaque-forming units assay
Plaque assays were performed on BHK-21 cells. In brief, BHK-21 monolayers were grown to 80% confluency in 24-well plates and incubated for 1 h at 37 °C in 5% CO2 with serially diluted virus supernatants from wild-type and mutant HAP1 cells infected with DENV, at a MOI of 0.1 for 48 h. The wells were then overlaid with DMEM, 0.8% Aquacide II (EMD Millipore), and 10% FBS, incubated for 7 days, and fixed with 10% formaldehyde. The cells were then stained overnight with crystal violet. The next day the wells were extensively washed with water then dried, and the resulting plaques were counted and plaque-forming units per ml were calculated. Two independent experiments were performed with triplicate infections and one representative is shown.
Focus-forming units assay
Wild-type and knockout Huh7.5.1 cells were plated in 24-well plates and infected with HCV at a MOI of 0.1. Three days after infection, supernatant was collected and added to wild-type Huh7.5.1 cells in a tenfold dilution series. After 3 days, cells were fixed, stained with mouse-anti-HCV-core (Abcam ab2740) and anti-mouse-IgG-Alexa-488 (Life Technologies) and fluorescent colonies were counted. Two independent experiments were performed with triplicate infections and one representative experiment is shown.
Luciferase reporter virus assays
Cells were plated out in 96-well plates in triplicates and infected with dengue luciferase reporter virus at an MOI of 0.01. Cells were incubated with dengue luciferase reporter virus at 37 °C, 5% CO2 and cell lysates were collected. Luciferase expression was measured using Renilla Luciferase Assay system (Promega E2820). Cells were lysed using Renilla lysis buffer and luciferase activity measured by addition of substrate and immediate luciferase readings were taken using Glomax 20/20 luminometer using a 10-s integration time. For the cross-comparison of the effects of host factors on DENV and HCV Huh7.5.1 knockout cell lines were infected with dengue luciferase reporter virus at an MOI of 0.01 or with HCV luciferase virus at an MOI of 0.2. For the validation of HCV host factors, four different knockout cell lines (created using lentiCRISPRv2) were infected with HCV luciferase virus at an MOI of 0.2. Two independent experiments were performed with triplicate infections and one representative experiment is shown, with the exception of the experiment shown in Extended Data Fig. 4e, which was performed once with triplicate infections.
Infection of Raji DC-SIGN cells
Raji DC-SIGN host factor knockout cell lines were created by transduction of lentiCRISPRv2 and subsequent puromycin selection. Resulting cell lines were infected with dengue luciferase virus at an MOI of 0.05 and collected 3 days after infection. Three independent experiments were performed with triplicate infections and one representative is shown.
Internalization assay and confocal microscopy
Approximately 10,000 Huh7 cells were seeded on Laboratory-TekII Chamber slides (Thermo Fisher Scientific). The next day, cells were incubated on ice for 15 min, infected with DENV (MOI = 60) and incubated on ice for 1 h. Cells were washed three times with ice-cold PBS and subsequently incubated at 37 °C for 0 or 30 min. At each time point, 10 μg ml−1 wheat germ agglutinin-Alexa-594 (Life Technologies, W11262) was added for 10 min at room temperature before three washes with PBS and fixation with 4% paraformaldehyde. Dengue virus was stained with a rabbit-anti-dengue-envelope antibody (Genetex, 127277) in block/perm buffer (1% saponin, 1% Triton X-100, 5% FBS) for 1 h followed by incubation with goat anti-rabbit-IgG-Alexa-488 (Life Technologies, A-11008) and DAPI (Insitus, F203) for 30 min. After three washes with PBS, cells were visualized using confocal microscopy.
Replicon assays
Dengue replicon plasmid was linearized using XbaI restriction enzyme. Replicon RNA was generated using the MEGAscript T7 High Yield Transcription Kit (Ambion, AM1334) with the reaction containing 5mM m7G(5′)ppp(5′)G RNA Cap Structure Analogue (NEB, S1405S). Resulting RNA was purified by sodium acetate ethanol precipitation. HCV sgJFH1 replicon33 RNA was prepared as described for DENV with the exception of adding the cap structure analogue. Cells were washed twice with PBS and re-suspended in electroporation buffer (Teknova, E0399). Three micrograms of purified replicon RNA was mixed with cells, and cells were electroporated using Bio-Rad Gene Pulser Xcell electroporator using square wave protocol. Electroporated cells were resuspended in cell culture medium without antibiotics and plated into 24-well plates. Luciferase expression was measured using Renilla Luciferase Assay system (Promega, E2820). Cells were lysed using Renilla lysis buffer and luciferase activity measured by addition of substrate and luciferase readings were taken immediately using Glomax 20/20 luminometer using a 10-s integration time. For lumiflavin treatment, cells were electroporated with 2 μg of viral RNA and 1 μg of firefly mRNA (Trilink) to normalize for effects on cell proliferation. For the lumiflavin treatment, two independent experiments with three electroporations each were performed. One representative experiment is shown. For the replicon assay in ELAVL1-knockout cells, three independent experiments with a single electroporation were performed. The average of the experiments is shown. For the DENV-replicon assay, three independent experiments were performed. One representative experiment was shown.
Immunoblot analysis
Cell pellets were lysed using Laemmli SDS sample buffer containing 5% β-mercaptoethanol and boiled for 10 min. Lysates were separated by SDS–PAGE on pre-cast Bio-Rad 4–15% poly-acrylamide gels in Bio-Rad miniprotean gel system. Proteins were transferred onto PVDF membranes using Bio-Rad trans-blot protein transfer system. PVDF membranes were blocked with PBS buffer containing 0.1% Tween-20 and 5% non-fat milk. Blocked membranes were incubated with primary antibody diluted in blocking buffer and incubated overnight at 4 °C rotating. Primary antibodies were detected using horseradish peroxidase (HRP)-conjugated secondary anti-mouse and anti-rabbit antibodies (Genetex) by incubating membranes at a 1:5,000 dilution for 1 h at room temperature. Antibody-bound proteins were detected by incubating with Pierce West Pico and Extended Duration Peroxide Solutions and visualized on film. Wild-type cells were treated with 10 μg ml−1 tunicamycin and treated for 3–4 h at 37 °C, 5% CO2. To visualize proteins by immunoblotting, the following antibodies were used anti-SHBG (Genetex, GTX63795) at a dilution of 1:2,500. Anti-pSAP (Genetex, GTX101064) at a dilution of 1:2,500. Anti-HA C29F4 (Cell Signaling, 3724P) at a dilution of 1:2,500. Anti-mouse M2-Flag (Sigma, F1804-200UG) at a dilution of 1:2,500. Anti-DYKDDDDK (Flag) (Cell Signaling, 2368) at a dilution of 1:2,500. Anti-RPN1 (gift from M. Ivessa) at a dilution of 1:2,000. Anti-NS1 (Genetex GTX124280) at a dilution of 1:2,500. Anti-P84 (Genetex GTX70220) at a dilution of 1:3,500. Anti-DENV-ENV (Genetex GTX127277) at a dilution of 1:2,500. Anti-prM (Genetex, GTX128092) at a dilution of 1:2,500. Anti-NS2B (Genetex, GTX124246) at a dilution of 1:2,500. Anti-NS3 (Genetex, GTX124252) at a dilution of 1:2,500. Anti-RFK (Sigma, SAB1409492) at a 1:500 dilution. Anti-FLAD1 (Santa Cruz Bio, sc-376819) at a 1:250 dilution. Anti-STT3B (Sigma, HPA036646) at a dilution of 1:1,000. Anti-MAGT1 (Proteintech Group, 17430-1-AP) at a dilution of 1:1,000. Anti-RPS25 (Abcam, 102940) at a dilution of 1:1,000. Anti-HUR (Santa Cruz, sc-5261) at a dilution of 1:200. Anti-SRRD (Sigma, HPA002945) at a dilution of 1:500.
Lentiviral or retroviral complementations
Lentiviral or retroviral transduction was used to create stable cell lines expressing a selected gene of interest. Respective genes of interest (see ‘Construction of lenti- or retroviral constructs’ section) were cloned into the pLenti-CMV-Puro-DEST vector (w118-1) (a gift from E. Campeau), or PMX-IRES-BLAST-DEST. Lentivirus or retrovirus produced in HEK293FT cells was used to transduce respective cell lines overnight. Cells stably expressing the gene of interest were selected by treatment with 1–4 μg ml−1 puromycin or 10–50 μg ml−1 blasticidin over 2 days (InvivoGen) along with untransduced cells as negative control.
Genome engineering
CRISPR guide RNA sequences were designed using the Zhang laboratory CRISPR design tool (see Extended Data Fig. 3 for CRISPR target sites). Corresponding oligonucleotides or geneblocks containing U6 promoter sequence and U6 termination sequence were ordered from IDT. Oligonucleotides were cloned into the Zhang laboratory generated Cas9 expressing pX458 guide RNA plasmid (Addgene) as previously described using Gibson assembly reaction New England Biolabs. Geneblocks were cloned into pCR-Blunt II-TOPO vector (Life Technologies). TOPO-cloned geneblocks were co-transfected into respective cells with a mCherry-expressing construct and hCas9-expressing vector (Addgene 41815 hCas9 Church pcDNA3.3-Topo) guide RNA encoded in the pX458 plasmids were transfected alone using Lipofectamine 2000 (Life technologies) according to manufactures guidelines. Transfected cells were single-cell sorted based on GFP or mCherry expression into 96-well plates using BD influx cell sorter. Clonal cell lines were allowed to expand and genomic DNA was isolated for sequenced based genotyping of targeted allele. For this, a 500–700-base pair (bp) region that encompassed the guide RNA-targeted site was amplified and the PCR product was Sanger sequenced. In haploid cells (HAP1), only one mutated allele was present in the sequenced PCR product and cellular subclones containing a frame shift mutations or large indels were selected. In aneuploid Huh7.5.1 cells, we regularly observed that the PCR product contained more than 1 trace, suggesting non-identical mutations in multiple alleles. In this case, the PCR product was cloned into a plasmid vector and colonies were sequenced to separate allele specific mutations. Subclones were chosen where all alleles were mutated. It should be noted that in aneuploid Huh7.5.1 cells, we sometimes observed cellular subclones where all mutant alleles contained the same mutation (for example, CD81 and ELAVL1). It has been reported that CRISPR/Cas9 technology can generate homozygous bi-allelic mutations more frequently than expected in diploid cells or cancer cells34,35, perhaps because both alleles were independently repaired in an identical manner or because one allele served as a template for homology-directed repair of the other allele. To create KO cell lines using lentiCRISPRv2 (Addgene) the following guide RNAs below were cloned into the vector, Huh7.5.1 cells were lentivirally transduced and selected with Puromycin. The following guide RNA sequences were used: ANKRD49: AGAAAGGAGTCTCCGCACTG; ANKRD49 guide2: ATGAACCGTTACGTCAAACC; ANKRD49 guide3: GCCCAAAGAAGCAATCTGCT; ANKRD49 guide4: AGAAAGGAGTCTCCGCACTG; CD81: GCGCCCAACACCTTCTATGT; CLDN1: CGATGGCGCCGATCCATCCC; ELAVL1: TTGGGCGGATCATCAACTCG; ELAVL1 guide2: TGTGAACTACGTGACCGCGA; ELAVL1 guide3: GGGCCTCCGAACCGTCGCGC; ELAVL1 guide4: AGAGCGATCAACACGCTGAA; EMC1: AGGCCGAATCATGCGTTCCT; EMC2: GATTGCCATTCGAAAAGCCC; EMC3: GTGCCACCTTCTCCTATGAC; EMC4: TGCTTGTCCAAGTAACCGAC; FKBPL: GTCAAGAAGATCGTAATCCG; FKBPL guide2: GAAGAGCCCGTCCATAGCAT; FKBPL guide3: ACAGAGCTAACTATGGGCGT; FKBPL guide4: GTTTCGGTAGGAGGGTCTCG; FLAD1: ACAGACCATTGAGACCTCCC; FLAD1 guide2: CATGCGCATCAACCCACTGC; FLAD1 guide3: TACAGGAGTAGGGGTCAGTC; FLAD1 guide4: TGTGTCCCTGGGGGTTGAAG; MAGT1: GAGCGAACATGGCAGCGCGT; MIR-122: GAGTTTCCTTAGCAGAGCTG; MMGT1: CAGGCACTTACGCTGCGCAG; non-targeting: GCCCAGACGCCCTAGAATAG; OCLN: ACGTAGAGTCCAGTAGCTGC; OSTC: TCAGTCATAGAACCGACACT; PPIA: GTACCCTTACCACTCAGTCT; RFK: TATCATGCATACCTTCAAAG; RFK guide2: GGTCAAGTGGTGCGGGGCTT; RFK guide3: CTATGGGGAAATCCTCAATG; RFK guide4: CCAACCATAGTAAATACCAG; RPN2: TCGCTACCACGTGCCAGTTG; SRRD: GACTGTTCTCAGTGAGAACG; SRRD guide2: GATAGATACCTTTGCAATGT; SRRD guide3: ATTGAAGTCCTTAACACCCT; SRRD guide4: AACAACTGAAGGCCCCTGTG; SSR2: CAATAGCAGGGGGATGCCGA; SSR3: GACCCTAGTAAGCACATATT; STT3A: GTACTCACGGATCAAACTCA; STT3B: TACAGCAAAAGAGTCTACAT; ZEB1: TGAAGACAAACTGCATATTG.
TALENs targeting AUP1 in HAP1 cells were generated as indicated in Extended Data Fig. 3. Cells were co-transfected with left and right TALEN-containing constructs and an mCherry-expressing construct using Lipofectamine 2000 (Life Technologies) according to manufacturer’s guidelines. Transfected cells were single-cell sorted based on mCherry expression into 96-well plates using BD influx cell sorter. Subclones were allowed to expand and genomic DNA was isolated for sequenced based genotyping of AUP1 allele. HAP1 cells containing gene-trap insertions in STT3A, STT3B, RPN1, SSR2, SSR3, ASCC2 and RPS25 were isolated by picking resistant colonies from the DENV-2 haploid genetic screen. Picked colonies were screened for gene-trap insertions using PCR with primers directed to the gene-trap and the flanking region of the gene of interest.
Co-immunoprecipitation
Wild-type HAP1 cells were transduced with STT3A–Flag, STT3B–Flag or RPS25–Flag lentiviral vectors (see ‘Construction of lenti- or retroviral constructs’ section). HAP1 cells expressing Flag-tagged proteins were trypsinized and washed once with PBS. Cells were lysed with TNM buffer (25 mM Tris-HCl, 15 mM NaCl, 5 mM MgCl2) containing 1% digitonin, 1 mM PMSF, and Halt Protease and Phosphatase Inhibitor Cocktail (Life Technologies, 78440) for 1 h on ice gently vortexing every 15 min. Cell lysates were clarified by centrifugation at 15,000g for 10 min. Clarified lysates were incubated at 4 °C overnight with Anti-FlagG M2 Magnetic Beads (Sigma M8823-5ml). Incubated beads were washed three times with TNM buffer containing 0.1% digitonin, 1 mM PMSF, 1× halt protease and phosphatase inhibitor. Cells were washed once with TNM buffer and competitively eluted with TNM buffer containing 150 ng μl−1 3× Flag Peptide (Sigma, F4799) for 30 min on ice. For immunoblotting, elutions were denatured by boiling in 5× sample buffer and analysed by SDS–PAGE using antibodies against DENV non-structural proteins. Elutions were also prepared for mass spectrometry analysis. Cells expressing RPS25–Flag, a host protein with a likely different mechanistic action, as well as untransduced cells, were used as a negative control in these experiments.
SYPRO ruby staining
After electrophoresis gel was fixed for 30 min in fixative buffer (50% methanol, 7% acetic acid) and incubated with SYPRO Ruby Stain (Fischer, S-12000) overnight. Gels were then washed once with wash buffer (10% methanol, 7% acetic acid) and twice with distilled water. Gel was imaged on Molecular Dynamics Storm scanner.
Construction of STT3A/STT3B double-knockout cell line with STT3B conditionally expressed using a Tet-On system
HAP1 cells stably transduced with the transactivator pLenti-CMVrtTA3G-Blast (R980-M38-658) (Addgene, 31797) with the endogenous STT3B gene disrupted by CRISPR/Cas9 were lentivirally transduced with pLenti-CMVTRE3G-Puro-STT3B-Flag, which drives STT3B under a doxycycline-inducible promoter. Transduced cells were then transfected with pX458 plasmid encoding a guide RNA targeted to the STT3A gene. Transfected cells were then subcloned based on GFP expression of PX458 plasmid into 96-well plates containing IMDM plus 10% FBS, penicillin-streptomycin, l-glutamine and 25 ng ml−1 doxycycline. Subclones were allowed to grow for 2 weeks, then replica plated and in one replicate the doxycycline medium was washed away and replaced with regular growth medium without doxycycline and incubated for 5 days. Cells that were dependent on doxycycline for growth were genotyped to verify both endogenous STT3A and STT3B had double frame shifting CRISPR/Cas9 editing events. STT3A/STT3B endogenous double knock out cells were then lentivirally transduced with wild-type or mutant STT3A or STT3B under the CMV promoter. The lentivirally transduced cell lines were then plated in 96-well plates and the doxycycline was washed away and incubated for 5 days. Cells were then fixed and stained with crystal violet to assess cell viability.
Treatment of HCV infected cells with lumiflavin, FMN and FAD
Wild-type Huh7.5.1 cells were treated with lumiflavin (Santa Cruz Bio, sc-224045) ranging from 10 to 100 μM and infected with HCV or DENV-2 at a MOI of 0.1. For WNV, YFV, PV (polio virus), SINV, VEEV and HRV-14 cells were treated with 50 μM lumiflavin and infected at a MOI of 0.1. For rescue of HCV replication in lumiflavin-treated cells 100 μM FMN and 10 mM FAD were used. RFK- and FLAD1-KO Huh7.5.1 were cultured in absence or presence of 500 μM FMN (TCI America, R0023) or FAD (Sigma, F8384) and subsequently infected with HCV at a MOI of 0.1. After 3 days of infection, levels of infection were determined using immunofluorescence, western blot and qPCR. Anti-HCV core 1b (Abcam ab2740) was used at 1:500 for immunofluorescence and 1:1,000 for western blotting. Anti-DENV-2 NS5 (GeneTex GTX103350) was used at a 1:25,00 dilution for western blotting. For the lumiflavin treatment two independent experiments with triplicate infections were performed and one representative is shown. For the FMN/FAD complementation two independent experiments were performed and the average is shown.
MTT assay
To test effects of lumiflavin on cell viability MTT assay was performed according to the manufacturer’s instructions (Sigma Cell Proliferation Kit I (MTT), 11465007001). Three independent experiments in triplicates each were performed and one representative is shown.
Transmission electron microscopy
Cells stably expressing STT3B-APEX2 were plated in 6-well plates and infected with an MOI 5 of DENV-2. 28 h after infection, cells were washed with PBS and fixed with 2% glutaraldehyde in 100 mM sodium cacodylate, 2 mM CaCl2, pH 7.4, buffer. Cells were fixed at 4 °C for 60 min then washed three times with PBS. Fixed cells were quenched with 100 mM sodium cacodylate, 2 mM CaCl2, pH 7.4, and 20 mM glycine. Quenched cells were washed twice with PBS and stained with using the KPL DAB reagent set (KPL, 54-10-00) for 8 h. After incubation with DAB, cells were rinsed twice with PBS and scraped off well using a cell scraper and pelleted. Pelleted cells were re-suspended in 10% gelatin in 0.1 M sodium cacodylate buffer, pH 7.4, at 37 °C and allowed to equilibrate for 5 min. Cells were pelleted again, excess gelatin removed, then chilled in cold blocks and covered with cold 1% osmium tetroxide (EMS, 19100) for 2 h rotating in a cold room. They were then washed three times with cold ultrafiltered water, then en bloc stained overnight in 1% uranyl acetate at 4 °C while rotating. Samples were then dehydrated in a series of ethanol washes for 20 min each at 4 °C beginning at 30%, 50%, 70%, 95% where the samples were then allowed to rise to room temperature, changed to 100% ethanol twice, then propylene oxide for 15 min. Samples were then infiltrated with EMbed-812 resin (EMS, 14120) mixed 1:2, 1:1 and 2:1 with propylene oxide for 2 h each with leaving samples in 2:1 resin to propylene oxide overnight rotating at room temperature in the hood. The samples were then placed into EMbed-812 for 2–4 h then placed into molds with labels and fresh resin, orientated and placed into a 65 °C oven overnight. Sections were taken at approximately 80 nm, picked up on formvar/carbon-coated 100-mesh Cu grids, stained for 30 s in 3.5% uranyl acetate in 50% acetone followed by staining in 0.2% lead citrate for 3 min. Observed in the JEOL JEM-1400 120kV and photos were taken using a Gatan Orius 4k × 4k digital camera.
Mass spectrometry
Liquid chromatography-tandem mass spectrometry
Elutions from co-immunoprecipitations were trypsin digested and purified using Sep Pak C18 purification column. Peptides were analysed using an LTQ Velos Orbitrap mass spectrometer (Thermo Fisher Scientific) coupled to an Agilent 1100 high performance liquid chromatography pump (Agilent Technologies) and a MicroAS autosampler (Thermo Fisher Scientific). Peptide mixtures were introduced into the mass spectrometer via a fused silica microcapillary column (100 μm inner diameter) ending in an in-house pulled needle tip (internal diameter ≈ 5 μm). Columns were packed to a length of 17 cm with a C18 reversed-phase resin (Magic C18AQ; Michrom Bioresources). Peptides were loaded onto the column and then eluted into the nanospray ionization source of the mass spectrometer via a two-step gradient of 7–25% buffer B (2.5% water and 0.1% formic acid in acetonitrile (v/v)) in buffer A (2.5% acetonitrile and 0.1% formic acid in water (v/v)) over 60 min followed by a second phase of 25–45% buffer B over 20 min. Eluting peptides were measured by the LTQ Velos Orbitrap operating in a data-dependent mode in which 10 ion-trap MS/MS spectra were acquired per data-dependent cycle from a high-resolution (R = 60,000) precursor spectrum (mass range = 360–1,600 m/z).
Mass spectrometry data processing
Raw data files produced by the mass spectrometer were converted to the mzXML format using in house software, MS Convert. MS and MS/MS data were extracted from mzXML files with in-house software. MS/MS spectra were analysed using Sequest algorithm searching a composite target-decoy protein sequence database. The target sequences consisted of human proteins downloaded from the Uniprot database (11-17-2014) and protein sequence corresponding to the dengue virus 2 16681 polyprotein. Decoy sequences were created by reversing the orientation of all target sequences. Parameters used for all searches included the requirement of trypsin peptide cleavage, two missed cleavages allowed, peptide mass tolerance of 20 p.p.m., variable oxidation of methionine residues (+15.99491 Da), and static carbamylation modification of cysteine residues (+57.02146). Decoy peptide identifications guided the creation of filtering criteria delivering preliminary sets of peptide-spectrum matches with estimated false discovery rate <1%. Spectral counts for each condition were combined at a protein level and normalized by protein length to infer protein abundances in each case.
DENV reporter virus and DENV replicon design and generation
Construction of pDENV-Luc replicon
The design of the DENV replicon was based on DVRep described previously36: The viral 5′ untranslated region (UTR) was followed by a duplication of the first 102 nucleotides of the C coding region, which contain cis-acting elements required for replication (CAE). The CAE was fused to the renilla luciferase coding region followed by the DENV open reading frame (ORF) starting at the signal peptide preceding NS1. Between the luciferase and the DENV structural proteins a foot and mouth disease virus (FMVD) 2A sequence was introduced to provide cotranslational cleavage and release of luciferase. The construct was based on pD2/IC-30P, which contains a full-length infectious clone encoding DENV-2 strain 16681 (ref. 37). We also included the amino acid mutation Q399H in the envelope protein. We gene-synthesized a fragment containing the T7 polymerase promoter sequence followed by the first 102 nucleotides of the C coding region in frame with Renilla luciferase and FMDV 2A followed by the DENV open reading frame (ORF) starting at the signal peptide preceding NS1 until an internal HpaI site. This fragment was released by SacI (preceding the T7 promoter) and HpaI and cloned in pD2/IC-30P in a three point ligation with KpnI/SacI and KpnI/HpaI fragments.
Construction of pDENV-Luc infectious clone
The design of the DENV reporter was based on mDV-R described previously38: The viral 5′ UTR was followed by a duplication of the first 104 nucleotides of the C coding region, which contain cis-acting elements required for replication (CAE). The CAE was fused to the renilla luciferase coding region followed by the complete DENV ORF. Between the luciferase and the DENV structural proteins a FMVD 2A sequence was introduced to provide cotranslational cleavage and release of luciferase. The construct was based on pD2/IC-30P, which contains a full-length infectious clone encoding dengue virus serotype 2 strain 16681 (ref. 37) in which an envelope Q399H mutation was introduced that enhanced viral infection in mammalian cells using primers 5′-GGAAGTTCTATCGGCCACATGTTTGAGACAAC-3′ and 5′-GTTGTCTCAAACATGTGGCCGATAGAACTTCC-3′ via the QuikChange Site-Directed Mutagenesis kit (Agilent Technologies). We gene-synthesized a fragment containing the T7 polymerase promoter sequence followed by the first 104 nucleotides of the C coding region in frame with Renilla luciferase and FMDV 2A. This fragment was PCR amplified, introducing a SacI site at the 5′ end and a NheI site (present in the FMDV 2A sequence) at the 3′ end using primers: 5′-CGAAATTCGAGCTCACGCG-3′ and 5′-TCCTGCTAGCTTGAGCAAATCAAAGTTC-3′. To create an in-frame fusion of FMDV 2A with the DENV ORF a second DNA fragment was amplified using pD2/IC-30P as template with primers: 5′-TCAAGCTAGCAGGAGACGTTGAGTCCAACCCCGGGCCCATGAATAACCAACGGAAAAAGGCG-3′ and 5′-GGAAGAGCATGCAGTCGGAAATG-3′ introducing 5′ NheI and 3′ SphI restriction sites. The two fragments were cut with the respective restriction enzymes and ligated into pD2/IC-30P cut with SacI and SphI to create pDENV-Luc. DENV-Luc virus was produced by cutting with XbaI to linearize plasmid and in vitro transcription performed of pDENV-Luc and transfection into BHK cells using Lipofectamine 2000.
Construction of lenti- or retroviral constructs
PMX-IRES-BLAST-DEST was made by cutting pMXs-IRES-Blasticidin Retroviral Vector (Cell Biolabs, RTV-016) with SnaBI and the Gateway destination cassette (reading frame A) was blunt cloned in to this vector according to manufacturer’s protocol (Gateway Vector Conversion System; Invitrogen, 11828-029)
To generate a lentiviral construct expressing STT3A–Flag, Dharmacon cDNA BC020965 was used as template to generate a PCR product using primers 5′-CACCATGACTAAGTTTGGATTTTTGCG-3′ and 5′-TTACTTATCGTCGTCATCCTTGTAATCTGTCCTTGACAAGCCTCGATT-3′
Amplified PCR product was then topo cloned into Gateway compatible entry vector pENTR/D-TOPO Cloning Kit (Life Technologies K2400-20) and gateway reaction (Life Technologies) used to insert into pLenti-CMV-Puro-Dest (w118-1).
To generate a lentiviral construct expressing STT3B–Flag, Dharmacon cDNA BC052433 was used as template to generate PCR product using primers forward primer 5′-CACCATGTCTTGGTGGGATTATGGC-3′ and reverse primer 5′-TTACTTATCGTCGTCATCCTTGTAATCAACAGTCTTCTTAGAGGTCTTCTT-3′. It should be noted that we used Mus musculus STT3B because we were unable to clone human STT3B.
Amplified PCR product was then TOPO cloned into Gateway compatible entry vector pENTR/D-TOPO Cloning Kit (Life Technologies, K2400-20) and gateway reaction was used to insert into pLenti-CMV-Puro-Dest (w118-1).
To generate an SHBG-expressing construct, SHBG was ordered as two geneblocks and used to generate PCR product with primers 5′-TGTGGTGGAATTCTGCAGATACCTGTGGTGGAATTCTGCAGATACC-3′ and 5′-ATCCAGCACAGTGGCGG-3′. PCR product was Gibson cloned Gibson assembly reaction kit (New England Biolabs) into pLenti-CMV-Puro-Dest (w118-1) that was EcoRV digested.
To generate a Flag3×–RPS25 expression construct, entry vector PENTR-Flag3×–RPS25 was generated as described39 using the forward primer CACCATGGACTACAAAGACCATGACGG. PENTR-Flag3×–RPS25 was then used in a Gateway reaction (Life Technologies) to introduce Flag3×–RPS25 into PMX-IRES-BLAST-DEST retroviral expression construct.
A construct expressing STT3B fused with APEX2 and Flag-tagged PCR products were generated using the pLenti-STT3B expression construct described above and APEX2 Addgene plasmid 49386 (refs 40, 41) as templates to generate PCR products using primers 5′-GACTCTAGTCCAGTGTGGTG-3′ with 5′-AACAGTCTTCTTAGAGGTCTTC-3′ and 5′-GAAGACCTCTAAGAAGACTGTTATGGACTACAAGGATGACGA-3′ with 5′-CGGCCGCCACTGTGCTGGATTTAGGCATCAGCAAACCCAAG-3′. PCR products were Gibson assembled Gibson assembly reaction kit (New England Biolabs) into pLenti-CMV-Puro-Dest (w118-1) that was EcoRV digested.
To generate a STT3B-doxycycline-lenti construct, Dharmacon cDNA BC052433 was used as a template to generate PCR product using primers 5′-CACCATGTCTTGGTGGGATTATGGC-3′ and 5′-TTACTTATCGTCGTCATCCTTGTAATCAACAGTCTTCTTAGAGGTCTTCTT-3′
Amplified PCR product was then TOPO cloned into Gateway compatible entry vector pENTR/D-TOPO Cloning Kit (Life Technologies, K2400-20), and gateway reaction was used to insert into the doxycycline inducible lentiviral vector pLenti CMVTRE3G Puro DEST (w811-1) (Addgene, 27565).
To generate catalytic site mutants (Extended Data Fig. 5 and refs 15, 42), STT3A- and STT3B-expressing constructs, DNA fragments were generated using pLenti-STT3B-Flag described above as a template using pLenti-EcoRV primers and mutant primers to generate two PCR products (see primers below). Both PCR products for each mutation were Gibson cloned (Gibson assembly reaction kit; New England Biolabs) into pLenti-CMV-Puro-Dest (w118-1) that was EcoRV digested.
Primers forward (F) and reverse (R) were as follows: pLenti EcoRV-F 5′-GACTCTAGTCCAGTGTGGTG-3′, pLenti EcoRV-R 5′-ATCCAGAGGTTGATTGTCGAG-3′. STT3A mutations: E63A-F 5′-CAGGTTCCTGGCTGAGGCCGGGTTTTATAAATTCCATAACTGG-3′, E63A-R 5′-CCGGCCTCAGCCAGGAACCTG-3′; D167A-F 5′-CTGTGGCTGGCTCCTATGCCAATGAAGGGATTGCCATCTTTTG-3′, D167A-R 5′-CATTGGCATAGGAGCCAGCCACAG-3′; E351Q-F 5′-CCATCATTGCTTCTGTGTCTCAGCATCAGCCCACAACCTG-3′, E351Q-R 5′-ATGCTGAGACACAGAAGCAATGATGG-3′. STT3B mutations: D100A-F 5′-ATCATCCACGAGTTCGCCCCGTGGTTTAACTATAG-3′, D100A-R 5′-CTATAGTTAAACCACGGGGCGAACTCGTGGATGAT-3′; D218A-F 5′-CAGTGGCGGGATCCTTTGCCAATGAAGGCATTGCCATT-3′, D218A-R 5′-AATGGCAATGCCTTCATTGGCAAAGGATCCCGCCACTG-3′; E402Q-F 5′-CAATTATTGCATCAGTGTCTCAGCATCAGCCTACGACATGG-3′, E402Q-R 5′-CCATGTCGTAGGCTGATGCTGAGACACTGATGCAATAATTG-3′.
ELAVL1 fused with Flag cDNA was prepared from total RNA of Huh7.5.1 cells using Biorad RT Superscript, and ELAVL1 was PCR amplified with 5′-TGTGGTGGAATTCTGCAGATACCATGTCTAATGGTTATGAAGACCA-3′ and 5′-CGGCCGCCACTGTGCTGGATTTACTTATCGTCGTCATCCTTGTAATCTTTGTGGGACTTG-3′. Next, using Gibson Assembly the PCR product was cloned into pLenti-CMV-Puro-Dest (w118-1) that was digested with EcoRV.
To generate RPN1 fused to 2Strep cDNA, BC010839 was PCR amplified with 5′-CACCATGGAGGCGCCAGCCGC-3′ and 5′-CTACAGGGCATCCAGGATG-3′. The amplified fragments were cloned into P-ENTR-D-Topo (Invitrogen) then a Gateway LR reaction was performed to shuttle cDNA into pLenti-CMV-puro expression vector.
To generate SSR2 fused to 2Strep cDNA, NM_003145.3 was PCR amplified with 5′-CACCATGAGGCTGCTGTCATTTGTG-3′ and 5′-TCAGTTCTTCTTCGTTTTGGGAG-3′. The amplified fragments were cloned into P-ENTR-D-Topo (Invitrogen) then a Gateway LR reaction was performed to shuttle cDNA into pLenti-CMV-puro expression vector.
To generate SSR3 fused to 2Strep cDNA, NM_003145.3 was PCR amplified with 5′-CACCATGGCTCCTAAAGGCAGCTC-3′ and 5′-CTATTTGGAGCCAGTAGACAG-3′. The amplified fragments were cloned into P-ENTR-D-Topo (Invitrogen) then a Gateway LR reaction was performed to shuttle cDNA into pLenti-CMV-puro expression vector.
To generate ASCC2 fused to 2Strep, BC025368 was PCR amplified with 5′-CACCATGCCAGCTCTGCCCCTGG-3′ and 5′-TCAGGATGGGATCATGCCTTTGCT-3′. The amplified fragments were cloned into P-ENTR-D-Topo (Invitrogen) then a Gateway LR reaction was performed to shuttle cDNA into pLenti-CMV-puro expression vector.
To generate RPS25 fused to 2Strep, NM_001028 was PCR amplified with 5′-CACCATGGACTACAAAGACCATGACG-3′ and 5′-TTAATTAACCTCGAGTTTAAACGCG-3′. The amplified fragments were cloned into P-ENTR-D-Topo (Invitrogen) then a Gateway LR reaction was performed to shuttle cDNA into pLenti-CMV-puro expression vector.
To generate the UBE2J1 expression construct, cDNA provided by R. Kopito was PCR amplified with 5′-TGTGGTGGAATTCTGCAGATACCATGGAGACCCGCTACAACCTG-3′ and 5′-CGGCCGCCACTGTGCTGGATTTATAACTCAAAGTCAAATATGTATTC-3′. The amplified fragments were cloned into pLenti-CMV-puro expression vector by a Gibson Assembly reaction.
To generate the SEL1L expression construct, cDNA provided by R. Kopito was PCR amplified with 5′-TGTGGTGGAATTCTGCAGATACCATGCGGGTCCGGATAGGGCTG-3′ and 5′-CGGCCGCCACTGTGCTGGATTTAAAGTCTACTTACCAAAACCATG-3′. The amplified fragments were cloned into pLenti-CMV-puro expression vector by a Gibson Assembly reaction.
To generate AUP1, glycerol stocks containing AUP1 cDNA in pENTR entry vector were ordered from Darmacon (OHS5894-99868092), and a Gateway LR reaction was performed to shuttle cDNA into pLenti-CMV-puro expression vector.
Comparison of knockout screens to siRNA screens
To compare the top 30 host factor genes of the knockout screens to results from previous short interfering RNA (siRNA) screens, we used the following data: (1) Sessions et al.27 (supplementary table 2). To rank the list by strength of phenotype, we used the P value. If multiple siRNA sequences per gene were present, we used the one with the stronger effect. (2) Krishnan et al.11 (supplementary table 1). To rank the identified DENV host factor, column AM was filtered for ‘required by both WNV and dengue’. The remaining genes were sorted by ‘pooled siRNA Fold reduction of DENV’ (column AN). (3) Tai et al.29 (table S2). To sort by phenotype, we chose the validated genes scoring with at least two siRNAs and ranked by P value. (4) Li et al.28 (from dataset S1). To rank the genes, we took the mean of the average normalized percentage infected cells part one or two of the four siRNAs. The top 10 genes based on phenotype as explained above are shown in Extended Data Fig. 10.
Accession codes
Primary accessions
BioProject
Data deposits
The CRISPR and haploid genetic screens have been deposited in the NCBI BioProject database under accession numbers PRJNA322191 and PRJNA284536, respectively.
Change history
15 July 2016
The HTML was replaced with the final print version of the manuscript (instead of the near-final version).
References
Bhatt, S. et al. The global distribution and burden of dengue. Nature 496, 504–507 (2013)
Thomas, S. J. & Rothman, A. L. Trials and tribulations on the path to developing a dengue vaccine. Am. J. Prev. Med. 49 (suppl. 4), S334–S344 (2015)
Lavanchy, D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 17, 107–115 (2011)
Paul, D. & Bartenschlager, R. Flaviviridae replication organelles: oh, what a tangled web we weave. Annu. Rev. Virol. 2, 289–310 (2015)
Shalem, O. et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014)
Wang, T., Wei, J. J., Sabatini, D. M. & Lander, E. S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84 (2014)
Rasmussen, S. A., Jamieson, D. J., Honein, M. A. & Petersen, L. R. Zika virus and birth defects—reviewing the evidence for causality. N. Engl. J. Med. 374, 1981–1987 (2016)
Carette, J. E. et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 326, 1231–1235 (2009)
Carette, J. E. et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343 (2011)
Fons, R. D., Bogert, B. A. & Hegde, R. S. Substrate-specific function of the translocon-associated protein complex during translocation across the ER membrane. J. Cell Biol. 160, 529–539 (2003)
Krishnan, M. N. et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 455, 242–245 (2008)
Ma, H. et al. A CRISPR-based screen identifies genes essential for West-Nile-Virus-induced cell death. Cell Rep. 12, 673–683 (2015)
Shrimal, S., Cherepanova, N. A. & Gilmore, R. Cotranslational and posttranslocational N-glycosylation of proteins in the endoplasmic reticulum. Semin. Cell Dev. Biol. 41, 71–78 (2015)
Ruiz-Canada, C., Kelleher, D. J. & Gilmore, R. Cotranslational and posttranslational N-glycosylation of polypeptides by distinct mammalian OST isoforms. Cell 136, 272–283 (2009)
Lizak, C., Gerber, S., Numao, S., Aebi, M. & Locher, K. P. X-ray structure of a bacterial oligosaccharyltransferase. Nature 474, 350–355 (2011)
Lindenbach, B. D. & Rice, C. M. trans-Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J. Virol. 71, 9608–9617 (1997)
Beatty, P. R. et al. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci. Transl. Med. 7, 304ra141 (2015)
Parnas, O. et al. A Genome-wide CRISPR screen in primary immune cells to dissect regulatory networks. Cell 162, 675–686 (2015)
Hasan, M. et al. Cytosolic nuclease TREX1 regulates oligosaccharyltransferase activity independent of nuclease activity to suppress immune activation. Immunity 43, 463–474 (2015)
Scheel, T. K. & Rice, C. M. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat. Med. 19, 837–849 (2013)
Jopling, C. L., Yi, M., Lancaster, A. M., Lemon, S. M. & Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309, 1577–1581 (2005)
Brennan, C. M. & Steitz, J. A. HuR and mRNA stability. Cell. Mol. Life Sci. 58, 266–277 (2001)
Sokoloski, K. J. et al. Sindbis virus usurps the cellular HuR protein to stabilize its transcripts and promote productive infections in mammalian and mosquito cells. Cell Host Microbe 8, 196–207 (2010)
Shwetha, S. et al. HuR displaces polypyrimidine tract binding protein to facilitate la binding to the 3′ untranslated region and enhances hepatitis C virus replication. J. Virol. 89, 11356–11371 (2015)
Lin, K. & Gallay, P. Curing a viral infection by targeting the host: the example of cyclophilin inhibitors. Antiviral Res. 99, 68–77 (2013)
Fujimura, M. et al. Functional characteristics of the human ortholog of riboflavin transporter 2 and riboflavin-responsive expression of its rat ortholog in the small intestine indicate its involvement in riboflavin absorption. J. Nutr. 140, 1722–1727 (2010)
Sessions, O. M. et al. Discovery of insect and human dengue virus host factors. Nature 458, 1047–1050 (2009)
Li, Q. et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl Acad. Sci. USA 106, 16410–16415 (2009)
Tai, A. W. et al. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 5, 298–307 (2009)
Ramage, H. R. et al. A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol. Cell 57, 329–340 (2015)
Sanjana, N. E., Shalem, O. & Zhang, F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014)
Luo, B. et al. Highly parallel identification of essential genes in cancer cells. Proc. Natl Acad. Sci. USA 105, 20380–20385 (2008)
Berger, K. L. et al. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl Acad. Sci. USA 106, 7577–7582 (2009)
Canver, M. C. et al. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem. 289, 21312–21324 (2014)
Horii, T. et al. Validation of microinjection methods for generating knockout mice by CRISPR/Cas-mediated genome engineering. Sci. Rep. 4, 4513 (2014)
Alvarez, D. E., De Lella Ezcurra, A. L., Fucito, S. & Gamarnik, A. V. Role of RNA structures present at the 3'UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 339, 200–212 (2005)
Kinney, R. M. et al. Construction of infectious cDNA clones for dengue 2 virus: strain 16681 and its attenuated vaccine derivative, strain PDK-53. Virology 230, 300–308 (1997)
Samsa, M. M. et al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 5, e1000632 (2009)
Fuchs, G. et al. Kinetic pathway of 40S ribosomal subunit recruitment to hepatitis C virus internal ribosome entry site. Proc. Natl Acad. Sci. USA 112, 319–325 (2015)
Lam, S. S. et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12, 51–54 (2015)
Martell, J. D. et al. Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat. Biotechnol. 30, 1143–1148 (2012)
Jaffee, M. B. & Imperiali, B. Exploiting topological constraints to reveal buried sequence motifs in the membrane-bound N-linked oligosaccharyl transferases. Biochemistry 50, 7557–7567 (2011)
Acknowledgements
The authors thank T. Brummelkamp, J. Idoyaga and S. Einav for critically reading the manuscript and discussions; X. Ji and the Stanford Functional Genomics Facility; The Stanford Transmission Electron Microscope Facility; The Stanford Shared FACS Facility, and members of the Carette laboratory for discussions and support. S. Einav, K. Kirkegaard, F. Chisari, J. F. Anderson, C. Rice, H. Ploegh, R. Kopito, E. Harris, M. Ivessa, S. Weaver, R. Tesh and E. Campeau are acknowledged for providing materials. The work was funded in part by National Institutes of Health (NIH) DP2 AI104557 (J.E.C.), NIH AI109662 (J.E.C.), David and Lucile Packard Foundation (J.E.C.), Stanford Graduate Fellowship (A.S.P.), Boehringer Ingelheim Fonds (A.S.P.) and NSF- GFRP (C.D.M.).
Author information
Authors and Affiliations
Contributions
C.D.M., A.S.P. and J.E.C. were responsible for overall design of the study. Most of the experiments related to the DENV and HCV genetic screens were performed by C.D.M. and A.S.P., respectively. K.M. designed and performed several validation experiments and J.E.C. designed dengue constructs. Y.S.O. performed one of the DENV screens. P.S. provided expertise in design of HCV experiments and M.A.M. and S.M.B. performed several HCV experiments. Mass spectrometry experiments were performed and analysed by C.D.M., G.F. and K.S. under the technical expertise of J.E.E. C.D.M., A.S.P., K.M. and J.E.C. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Additional information
Reviewer Information
Nature thanks W. Wei and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Figure 1 Divergence of DENV and HCV host factors.
a, Gene Ontology (GO) analysis for DENV and HCV CRISPR screens on the ranked gene lists. Curated (by redundancy) enriched GO terms are shown. A complete list of all enriched GO terms can be found in Supplementary Table 4. b, Distribution of the subcellular location of the 30 most enriched host factors for DENV and HCV. c, Cross-comparison of the effects of DENV or HCV host factor knockout in Huh7.5.1 cells on the replication of DENV or HCV using reporter viruses expressing luciferase. Data are mean and s.d. for triplicate infections.
Extended Data Figure 2 Reproducibility of CRISPR screens.
a, Ranked lists of the 30 most enriched DENV and HCV host factors and their rankings in the individual replicate screens. The colour code reflects in what percentile the gene scored in the replicate. b, Gene enrichment based on RIGER score for the individual replicate screens. Red dots highlight where the 30 most significant host factors ranked in the individual replicates.
Extended Data Figure 3 Genotyping of cell lines for DENV host factors.
a, The site of gene-trap insertion in HAP1 cell lines was determined using a PCR-based method. Bases depicted in red are the flanking sequences upstream of the gene-trap insertion. Bases depicted in green are downstream gene-trap flanking sequences. b, TALENs were used to edit the genomic region of AUP1 in HAP1 cells. Bases depicted in red are the left TALEN-binding site, bases depicted in blue are the right TALEN-binding site. Bases depicted in green are the TALEN target site. Arrow indicates site of 260-bp insertion. c, CRISPR Cas9 nuclease was targeted to bases depicted in red in HAP1 cells. Editing events are depicted at the guide RNA target sites below the wild-type sequence. d, CRISPR Cas9 nuclease was targeted to bases depicted in red in Huh7 cells. Editing events are depicted at the guide RNA target sites below the wild-type sequence. e, Immunoblots of wild-type and knockout cell lines.
Extended Data Figure 4 Validation of DENV host factor genes.
a, Plaque-forming units (PFU) assay of DENV infection. ND, no plaques detected (threshold of detection is 6 PFU ml−1). b, DENV luciferase levels in HAP1 isogenic knockout cells complemented using lentiviral stable expression of corresponding genes. c, Crystal violet of complemented Huh7 knockout cells infected with DENV. d, DENV luciferase levels in Raji DC-SIGN cells with knockout in DENV host factors (lentiCRISPRv2). Empty denotes an empty vector control (expressing Cas9 but no guide RNA), and NT denotes a cell line expressing a non-targeting guide RNA. e, Time course of DENV and HCV expressing Renilla luciferase in Huh7 knockout cells. f, Schematic diagram of the STT3A and STT3B isoforms. Gene names in red indicate OST subunits identified in the DENV screens. Data are mean and s.d. for triplicate infections.
Extended Data Figure 5 Catalytic site mutations introduced in mammalian STT3A and STT3B.
a, Catalytic site amino acids highlighted in red as identified in the bacterial STT3 (Campylobacter lari pglb). Strong conservation allows their identification in other species. Alignments of STT3 isoforms across different species highlight the conserved catalytic sites that were mutated. The table specifies the amino acid position and the specific triple mutations that were made to abolish catalytic activity. b, Huh7 STT3A and STT3B knockout cells expressing Flag-tagged STT3A and STT3B wild-type and catalytic mutants.
Extended Data Figure 6 Physical interaction between the OST complex and the replication complex of DENV.
a, APEX2, a protein tag for electron microscopy, was fused to the C terminus of STT3B enabling the imaging of subcellular protein localization by deposition of a polymer of 3,3′-diaminobenzidine (DAB). b, Luminescence of Huh7 STT3B-knockout cells complemented with STT3B–APEX2 and infected with DENV expressing Renilla luciferase. Data are mean and s.d. for triplicate infections. c, STT3B localizes on ER membranes in the vicinity of DENV-induced vesicle packets as shown by transmission electron microscopy micrograph of DENV-infected or uninfected Huh7 cells expressing the STT3B–APEX2 construct. N represents the cell nucleus, and the arrowheads in samples transfected with STT3B–APEX2 represent APEX polymerized DAB staining in the lumen of the ER or around DENV-induced vesicle packets (VP). d, Co-immunoprecipitations of STT3A–Flag and STT3B–Flag from DENV-infected cell lysates. LE, long exposure. e, Anti-Flag western blots of immunoprecipitation elutions of DENV-infected cells stably expressing Flag-tagged STT3A, STT3B and RPS25. f, SYPRO Ruby staining of elutions and inputs of immunoprecipitations of DENV-infected cell lysates. g, Co-immunoprecipitation elutions of DENV-infected lysates were analysed by mass spectrometry and DENV-specific peptides aligned to DENV polyprotein.
Extended Data Figure 7 Analysis of HCV host factor knockout cell lines.
a, Genotyping of CRISPR-induced knockout Huh7.5.1 cells by Sanger sequencing showing the mutated locus and the wild-type reference. CRISPR/Cas9 induces mutations close to the PAM site resulting in frameshifts. CD81 and ELAVL1 knockout cell lines are subclones, whereas others are populations of cells mutagenized with lentiCRISPRv2. b, Immunoblots of CRISPR-induced knockout cells.
Extended Data Figure 8 ELAVL1 is a critical host factor for HCV replication.
a, HCV luciferase infection in knockout cell lines using four different guide RNAs per gene. NT, non-targeting guide RNA. b, qPCR of viral RNA in wild-type or ELAVL1-knockout Huh7.5.1 cells. c, Crystal violet assay for different RNA virus infections. d, HCV replicon assays using wild-type sgJFH1 (left) or GND sgJFH1 replicon. Note that one downward error bar for the right panel (6 hours, ELAVL1-KO) was not plotted because it would reach a negative value, which cannot be plotted on a logarithmic scale. e, Transfection of ectopically expressed ELAVL1 restores HCV replication. Western blot of ELAVL1–Flag transfected and untransfected Huh7.5.1 ELAVL1-knockout cells. Data are mean and s.e.m. (qPCR) or s.d. (FFU, RLU) for triplicate infections, except in panel e, which was a single infection.
Extended Data Figure 9 Lumiflavin inhibits the replication of HCV but not of other RNA viruses.
a, qPCR of HCV or DENV RNA replication in wild-type, RFK-knockout or FLAD1-knockout Huh7.5.1 cells. b, Immunofluorescence of HCV infection in wild-type, RFK-knockout and FLAD1-knockout Huh7.5.1 cells under treatment with lumiflavin, FMN or FAD. HCV core protein (green). Blue denotes DAPI (nuclear) staining. Scale bar, 57 μm. c, Western blot for HCV core and DENV NS5 in untreated (UT) and lumiflavin-treated Huh7.5.1 cells. p84 and GAPDH served as loading controls. d, qPCR of RNA viruses in untreated or lumiflavin-treated Huh7.5.1 cells. e, MTT cell proliferation assay for lumiflavin-treated Huh7.5.1 cells. f, Restoration of HCV replication in lumiflavin-treated cells by exogenous addition of FMN or FAD. Data are mean and s.e.m. (qPCR) or s.d. (MTT) for triplicate infections/treatments.
Extended Data Figure 10 Comparison of knockout screen results to previous siRNA screens.
a, Venn diagram comparing the hits from the CRISPR and haploid screens for DENV host factors to previous siRNA screens from Sessions et al.27 (from supplementary table 2) and Krishnan et al.11 (from supplementary table 1). The top ten validated host factors (by strength of phenotype in the validation screen) for each screen are shown next to the circle. b, Venn diagram comparing the hits from the CRISPR screen for HCV host factors to previous siRNA screens from Tai et al.29 (from table S2) and Li et al.28 (from dataset S1).
Supplementary information
Supplementary Table 1
This file contains Supplementary Table 1, Haploid screen DENV. Ranked table of haploid screen for DENV host factors showing gene name, number of independent inactivating gene-trap insertions in the selected cells, total number of inactivating gene-trap insertions in selected cells, number of independent gene-trap insertions in the unselected control cells, total number of gene-trap insertions in unselected cells, p-value of one-sided Fisher exact test and FDR-corrected p-value. (XLSX 340 kb)
Supplementary Table 2
This file contains Supplementary Table 2, CRISPR screen DENV. Ranking of DENV host factor genes by their mean RIGER score calculated from the average of 3 independent experiments. The results of the replicate screens are shown in tabs and display the following: Gene name, guideRNAs, number of guideRNAs in the library, enrichment rank of the individual guideRNAs, normalized enrichment score (RIGER score), gene rank, p-value and the number of guides scoring in the top 500, 1000, 5000, 10000. (XLSX 8566 kb)
Supplementary Table 3
This file contains Supplementary Table 3, CRISPR screen HCV. Ranking of HCV host factor genes by their mean RIGER score calculated from the average of 3 independent experiments. The results of the replicate screens are shown in tabs and display the following: Gene name, guideRNAs, number of guideRNAs in the library, enrichment rank of the individual guideRNAs, normalized enrichment score (RIGER score), gene rank, p-value and the number of guides scoring in the top 500, 1000, 5000, 10000. (XLSX 8586 kb)
Supplementary Table 4
This file contains Supplementary Table 4, Gene ontology and cellular compartment analysis. Gene ontology analysis (Gorilla) for biological process, function and component of the ranked gene lists obtained from the CRISPR screens. Analysis of subcellular localization of the top 30 DENV and HCV host factors. (XLSX 100 kb)
Supplementary Table 5
This file contains Supplementary table 5, Co-immunoprecipitation of host and DENV2 proteins in STT3A-FLAG or STT3B-FLAG pull down experiments identified by mass spectrometry analysis. Spectral counts and protein identifying information for high confidence peptides (<1% FDR) are shown for the indicated FLAG pulldown experiments. DENV2 specific peptides are shown in separate sheets. Known oligosaccharyltransferase interacting proteins are highlighted in yellow. (XLSX 77 kb)
Supplementary Figure 1
This file contains raw source data for immunoblots (Fig. 3b, c, e and Extended Data Fig. 3e, 5b, 6d, e, 7b, 8e, 9c) and protein gel (Extended Data Fig. 6f). (PDF 2072 kb)
Rights and permissions
About this article

Cite this article
Marceau, C., Puschnik, A., Majzoub, K. et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535, 159–163 (2016). https://doi.org/10.1038/nature18631
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature18631
This article is cited by
-
Host E3 ligase Hrd1 ubiquitinates and degrades H protein of canine distemper virus to inhibit viral replication
Veterinary Research (2023)
-
The non-classical major histocompatibility complex II protein SLA-DM is crucial for African swine fever virus replication
Scientific Reports (2023)
-
Hepatitis C virus RNA is 5′-capped with flavin adenine dinucleotide
Nature (2023)
-
Cellular glycan modification by B3GAT1 broadly restricts influenza virus infection
Nature Communications (2022)
-
Genome-wide CRISPR screen for HSV-1 host factors reveals PAPSS1 contributes to heparan sulfate synthesis
Communications Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
