
Abstract
Early T-cell precursor acute lymphoblastic leukaemia (ETP ALL) is an aggressive malignancy of unknown genetic basis. We performed whole-genome sequencing of 12 ETP ALL cases and assessed the frequency of the identified somatic mutations in 94 T-cell acute lymphoblastic leukaemia cases. ETP ALL was characterized by activating mutations in genes regulating cytokine receptor and RAS signalling (67% of cases; NRAS, KRAS, FLT3, IL7R, JAK3, JAK1, SH2B3 and BRAF), inactivating lesions disrupting haematopoietic development (58%; GATA3, ETV6, RUNX1, IKZF1 and EP300) and histone-modifying genes (48%; EZH2, EED, SUZ12, SETD2 and EP300). We also identified new targets of recurrent mutation including DNM2, ECT2L and RELN. The mutational spectrum is similar to myeloid tumours, and moreover, the global transcriptional profile of ETP ALL was similar to that of normal and myeloid leukaemia haematopoietic stem cells. These findings suggest that addition of myeloid-directed therapies might improve the poor outcome of ETP ALL.
Similar content being viewed by others


Main
Acute lymphoblastic leukaemia (ALL) is the most common malignancy of childhood, with 85% of cases being of B-cell lineage, and 15% T-cell lineage1. Recent studies have identified a subtype of T-cell acute lymphoblastic leukaemia (T-ALL) termed “early T-cell precursor” (ETP) ALL that comprises up to 15% of T-ALL, and is associated with a high risk of treatment failure2. ETP ALL is characterized by lack of expression of the T-lineage cell surface markers CD1a and CD8, weak or absent expression of CD5, aberrant expression of myeloid and haematopoietic stem cell markers (for example, CD13, CD33, CD34 and CD117), and a gene expression profile reminiscent of the murine early T-cell precursor3. The normal ETP, or double negative 1 (DN1) thymocyte retains the ability to differentiate into cells of both the T-cell and myeloid, but not B-cell, lineages4.
Somatic genetic alterations in ETP ALL
ETP ALL cases commonly exhibit a high burden of DNA copy number alterations, but lack a known unifying genetic alteration2. To define the landscape of genetic alterations in ETP ALL, The St Jude Children's Research Hospital–Washington University Pediatric Cancer Genome Project performed whole-genome sequencing (WGS) for matched leukaemic and normal DNA from 12 children with ETP ALL (Supplementary Tables 1–4 and 7, Supplementary Figs 2–4), and determined the frequency of mutations in a separate cohort of 52 ETP and 42 non-ETP childhood T-ALL cases, 82 of which had matched remission DNA. Transcriptome sequencing was performed for two WGS cases, and whole-exome sequencing for three ETP samples in the recurrence cohort (Supplementary Table 8). Putative somatic sequence mutations, and structural alterations identified using CREST5, were validated by PCR and sequencing. We identified an average of 1,140 sequence mutations (range 235–1,929) and 12 structural variations (range 0–25) per case (Supplementary Tables 9, 11 and 12, Supplementary Fig. 5), including 154 non-silent sequence variants. Fifty-four per cent of the missense mutations were predicted to be deleterious (Supplementary Table 12), indicating that many of these variants are involved in leukaemogenesis.
Structural rearrangements in ETP ALL
We detected 181 structural variations across the WGS cases (Supplementary Results and Supplementary Tables 13, 14, Fig. 1 and Supplementary Fig. 6). Most abnormalities identified by cytogenetics were also evident on analysis of WGS data (Supplementary Table 15). We also observed evidence of telomere shortening on analysis of WGS data (Supplementary Fig. 7). Three cases (SJTALL001, SJTALL002, SJTALL003) had multiple complex rearrangements with breakpoints suggestive of a single cellular catastrophe (“chromothripsis”6; Supplementary Figs 6 and 8). Case SJTALL001 had a nonsense mutation in MLH3, a DNA mismatch repair gene with a role in DNA double strand break repair, and SJTALL003 had a missense mutation in DCLRE1C, which encodes the non-homologous end-joining factor ARTEMIS, indicating a potential causal relationship between these mutations and the acquisition of structural variations. Case SJTALL007 had a deletion disrupting the mismatch repair gene MSH5, and also harboured multiple structural rearrangements.

Loss-of-heterozygosity, orange; amplification, red; deletion, blue. Sequence mutations in RefSeq genes: silent single-nucleotide variants (SNVs), green; non-silent SNVs, brown; indels, red. Genes at structural variant breakpoints: genes involved in in-frame fusions, pink; others, blue. Circos plots for all cases are provided in Supplementary Fig. 6.
Remarkably, 51% (77 out of 151) of the validated structural variations had breakpoints in coding genes, including genes with known roles in haematopoiesis and leukaemogenesis, or genes also targeted by sequence mutations (for example, MLH3, SUZ12 and RUNX1). A majority of these structural variations (65 out of 77, 84%) are predicted to result in loss-of-function of the involved genes, or occur as part of complex translocations that result in the formation of chimaeric fusion proteins. Ten chimaeric genes encoding six fusion proteins were detected in five cases (Supplementary Table 16) which resulted in the expression of chimaeric in-frame novel fusion genes disrupting haematopoietic regulators, including ETV6–INO80D (case SJTALL002), NUP214–SQSTM1 (SJTALL009) and NAP1L1–MLLT10 (SJTALL013) (Supplementary Figs 9–12). Case SJTALL012 harboured a RUNX1–EVX1 rearrangement arising from transplicing (SJTALL012; Supplementary Figs 10 and 11). No additional cases with these chimaeric fusions were identified upon testing 77 ETP and non-ETP ALL cases with available RNA by PCR with reverse transcription. However, exome sequencing identified ETV6–INO80D in case SJTALL208 (Supplementary Fig. 13). ETV6 encodes a transcription factor required for definitive haematopoiesis that is frequently altered in leukaemia7,8,9. Deletions and mutations of ETV6 were present in 33% of ETP and 10% of non-ETP T-ALL cases (Supplementary Fig. 14).
Sequence mutations in ETP ALL
In addition to genes known to be mutated in T-ALL, including NRAS10,11 (N = 3 out of 12 WGS cases) JAK1 (ref. 12, N = 2), NOTCH1 (ref. 13, N = 1), FLT3 (refs. 14–16, N = 1), PHF6 (ref. 17, N = 3) and WT1 (ref. 18, N = 1), (Supplementary Fig. 15), we identified multiple novel recurring targets of mutation. These included DNM2 (N = 2), ECT2L (N = 2), EP300 (N = 2), GATA3 (N = 2), IL7R (N = 2), JAK3 (N = 3), RELN (N = 2) and RUNX1 (N = 4) (Table 1, Fig. 2, Supplementary Tables 17 and 18, Supplementary Fig. 15). For the two cases also analysed by transcriptome sequencing (SJTALL002 and SJTALL012), 21 out of 38 mutations were expressed. We did not observe selective expression of mutant alleles, with the exception of those with a concomitant deletion of the wild-type allele (for example, KRAS in SJTALL002).

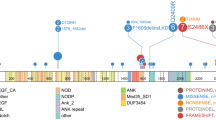
Recurring mutations in ETP-ALL. The figures show mutations for the 12 WGS cases, and the recurrence cohort of 94 cases sequenced by Sanger sequencing. The majority of cases had matched remission DNA to distinguish somatic from inherited variants. Where remission DNA was not available, but variants are known or predicted to be deleterious, mutations are shown as ‘variants’. The results of recurrence screening for additional genes sequenced are shown in Supplementary Table 17 and Supplementary Figs 14–17 and 20). The schematics are based on the following NCBI protein reference sequences: GATA3, NP_001002295; DNM2, NP_001005360; ECT2L, NP_001071174; EZH2, NP_001190176; PHF6, NP_001015877.1 and RUNX1, NP_001745.
Of 42 genes analysed by Sanger sequencing and single-nucleotide polymorphism microarray analysis in the recurrence cohort, 27 were recurrently mutated (Supplementary Table 19, Figs 2 and 3a, and Supplementary Figs 15 and 16). Of 254 validated non-silent mutations (Supplementary Table 17), 40.7% were indel mutations and 9.4% were nonsense mutations. Eighty-two per cent of missense mutations were predicted to be deleterious, a marked enrichment compared with mutations identified in the WGS samples, consistent with the majority being driver mutations.

a, Data are shown for 106 T-ALL cases, including the 12 cases that were subjected to whole-genome sequencing (arrowed), and 94 recurrence cases (52 ETP ALL and 42 non-ETP T-ALL). Cases have been grouped by ETP status, and cases lacking any mutations are shown to the left, followed by cases with NRAS and FLT3 mutations. Genes identified as novel targets of mutation in T-ALL are labelled in green. Four cases died while in remission and are excluded from outcome analysis. b, Frequency of somatic alterations targeting haematopoietic and lymphoid development in ETP and non-ETP T-ALL, showing an increased frequency of lesions in these pathways in ETP ALL. ***P < 0.0001.
We observed a high frequency of mutations known or predicted to result in aberrant cytokine receptor and RAS signalling in ETP ALL. Forty-three out of 64 (67.2%) of ETP cases had mutations in these pathways, compared to 8 out of 42 (19%) non-ETP cases (P < 0.0001; Table 1, Fig. 3b and Supplementary Table 20). Known or predicted activating mutations were identified in BRAF, FLT3, IGFR1, JAK1, JAK3, KRAS and NRAS (Supplementary Results). Three cases harboured the JAK3 M511I mutation located adjacent to the pseudokinase domain that has been identified previously in acute myeloid leukaemia and is transforming when introduced into murine haematopoietic progenitor cells19. The pseudokinase domain mutation, A573V, has previously been identified in acute megakaryoblastic leukaemia and is transforming20. The mutations identified in JAK1 are novel, but are in close proximity to sites of activating mutations previously identified in ALL12.
Seven cases (five ETP and two non-ETP) harboured mutations in IL7R encoding the IL7RA (interleukin 7 receptor alpha) chain (Fig. 4a). IL7RA forms a heterodimer with IL2RG (common gamma chain) for the cytokine IL7, and with CRLF2 (cytokine receptor like factor 2) forms a receptor for TSLP (thymic stromal lymphopoietin). IL7R and CRLF2 signalling are important in early lymphoid maturation21. Rearrangement of CRLF2 is observed in B-progenitor ALL22,23, and IL7R mutations have recently been identified in ALL24. All seven cases had an in-frame insertion or substitution at residues I241–V253 of the IL7R transmembrane domain. Consistent with prior data, expression of several of the IL7R mutant alleles in the cytokine-dependent murine haematopoietic Ba/F3 and MOHITO25 cell lines resulted in transformation to cytokine-independent cell growth (Fig. 4b, c). In six cases the mutations introduced a cysteine into the transmembrane domain that induces dimerization of the receptor in the absence of ligand (Fig. 4d). The mutations also induced Stat5 phosphorylation that was attenuated by Jak inhibition (Fig. 4e). Expression of mutant, but not wild type Il7r in primary murine haematopoietic progenitors resulted in enhanced colony replating in vitro (Fig. 4f, g), indicating that the IL7R alterations are transforming events in T-ALL.

a, Domain structure of IL7R, showing two hotspots of missense and in-frame insertion-deletion mutations (IL241-242 and VA253-254). The single case with a mutation in the amino-terminal region (V78M) is accompanied by a transmembrane domain mutation. b, c, Murine Il7r mutant alleles homologous to the human IL7R mutations were expressed in the murine haematopoietic IL-3-dependent Ba/F3 cell lines (b) or the murine IL-7-dependent MOHITO T-ALL cell line (c). WT, wild type. Expression of these mutant alleles resulted in transformation to cytokine-independent proliferation. MIG, empty MSCV-IRES-GFP vector. In b, growth curves have been offset to permit visualization of each allele. Error bars represent mean ± s.d. for three replicates. In c, transformation to cytokine-independent growth is shown as an increasing proportion of Il7r-mutant-expressing cells (or as a positive control, BCR–ABL1), as measured by the percentage of GFP-positive cells. d, Western blotting for Il7r in MOHITO cells transduced with wild-type or mutant Il7r alleles, showing the formation of Il7r dimers in cells expressing mutant alleles with an unpaired cysteine residue. e, Phosphosignalling analysis of MOHITO cells transduced with MIG, WT Il7r or four different Il7r mutant alleles, showing increased Stat5 phosphorylation in cells stimulated with IL7, or cells expressing mutant Il7r alleles in the absence of cytokine. Stat5 phosphorylation was reduced following exposure to Jak inhibitor I (inh) at 3 μM for 1 hour. f, g, Clonogenic assays of lineage-negative WT or Arf−/− murine bone marrow cells expressing mutant Il7r alleles show enhanced replating compared to cells transduced with empty vector (f), and enhanced replating compared to cells expressing WT Il7r cultured in the absence of IL7 (g). Columns show mean of two replicates ± s.e.m.
We also identified a high frequency of alterations of genes with roles in haematopoietic and lymphoid development, including RUNX1, IKZF1, ETV6, GATA3 and EP300 (57.8% of ETP cases versus 16.7% of non-ETP T-ALL cases, P < 0.0001). Importantly, several of these genes were targeted by multiple mechanisms of alteration across the cohort: sequence mutation, deletion and chromosomal translocations. Six cases (all ETP) had inactivating mutations of GATA3, four of which were biallelic due to either biallelic sequence mutations (SJTALL179, R276Q and A310_T317>VRP; SJTALL010 N286T and S271_W275fs) (Fig. 2) or due to concomitant deletion of the second allele (Supplementary Table 18). GATA3 encodes GATA binding protein 3, a member of a family of highly conserved zinc-finger transcription factors that is required for the development of early T-lineage progenitors26, and is mutated in the hypoparathyroidism with sensorineural deafness and renal dysplasia syndrome (HDR)27. In four cases the mutation was at R276, a residue also mutated in HDR27. The R276P mutation results in impaired DNA-binding affinity of GATA3 for its DNA targets, indicating that the mutations observed in T-ALL are likely to be loss of function. An additional case, SJTALL011, harboured a somatic mutation in GATA2, R307W, which is also located in the highly conserved GATA zinc-finger domain and is homologous to the GATA3 R276W mutation.
Twelve cases (ten ETP, and two non-ETP) harboured alterations of RUNX1. Two cases had concomitant deletion of the non-mutated allele, and three had RUNX1 deletions but no sequence mutation. RUNX1 is required for definitive haematopoiesis28 and normal T-lymphoid development, and is commonly rearranged and mutated in myeloid and lymphoid malignancies (Supplementary results)8,29,30,31,32. The mutations observed in T-ALL commonly involve the Runt domain, include frameshift and nonsense mutations, and are predicted to be deleterious. Nine cases (eight ETP) had deletions or sequence mutations of IKZF1 (IKAROS), which encodes a zinc-finger transcription factor required for the development of all lymphoid lineages that is commonly mutated in high-risk B-progenitor ALL and murine models of T-ALL (Supplementary Fig. 16).
A notable finding was a high frequency of somatic alterations targeting histone modification in ETP ALL. Six WGS cases had alterations in genes encoding components of the polycomb repressor complex 2 (PRC2), including deletions and sequence mutations of EED, EZH2 and SUZ12 (Table 1, Fig. 2 and Supplementary Fig. 17). EZH2 catalyses trimethylation of histone 3 lysine 27 (H3K27), resulting in transcriptional repression of genes involved in development, stem cell maintenance and differentiation33. Twenty-seven (42.2%) of ETP ALL cases harboured a deletion and/or sequence mutation in these genes, compared to five (11.9%) of non-ETP T-ALL cases (P = 0.001). Gain-of-function EZH2 Y641 mutations are common in lymphoma34. In contrast, structural modelling predicts that the mutations observed in T-ALL are likely to disrupt the catalytic SET domain and result in loss of function (Supplementary Results and Supplementary Figs 18 and 19). In addition, case SJTALL192 harboured a focal homozygous deletion of SETD2 which encodes a H3K36 trimethylase, and an additional four cases had loss-of-function mutations of this gene (Supplementary Fig. 20). Three cases had predicted loss-of-function mutations of the histone acetyltransferase gene EP300 (p300). Together, 31 ETP and 5 non-ETP cases had mutations affecting epigenetic regulation, which were biallelic or involved multiple genes in 10 cases (9 ETP and 1 non-ETP).
Novel recurrent somatic mutations
Recurring mutations were also identified in genes not previously known to be involved in lymphoid development or oncogenesis. DNM2 was mutated in 17 cases (13 ETP, 4 non-ETP), including two cases with biallelic mutations (Fig. 2). DNM2 encodes dynamin 2, a member of a family of large GTPases, and is involved in a wide range of cellular functions, including endocytosis, phagosome formation, intracellular trafficking, interaction with the actin and microtubule networks, and promotion of apoptosis35. Inherited DNM2 mutations result in the degenerative neurologic diseases Charcot–Marie–Tooth peripheral neuropathy and autosomal dominant centronuclear myopathy35. As in these diseases, the mutations in T-ALL are located throughout the gene in each functional domain, and include missense, nonsense, splice site and frameshift mutations, and are therefore likely to result in loss of DNM2 function. The role of DNM2 in lymphoid development and tumorigenesis is unknown, although it is expressed in leukaemic lymphoblasts (Supplementary Fig. 22).
Eight cases had missense, nonsense or splice site mutations in ECT2L (epithelial cell transforming sequence 2 oncogene gene like). Four cases had non-synonymous mutations in RELN, which encodes reelin, a large secreted extracellular matrix protein involved in the regulation of neuronal migration, and which is mutated in the neurodevelopmental disorder autosomal recessive lissencephaly with cerebellar hypoplasia36. Notably, several cases had inherited mutations in these two genes that are predicted to be deleterious. Sequence mutations were also found in 12 regulatory RNA genes including one microRNA gene (MIR1297).
Mutations in multiple pathways in ETP ALL
Recurring mutations targeting genes regulating haematopoietic development (‘type II lesions’, for example, GATA3, RUNX1, ETV6, IKZF1 and EP300) and cytokine receptor and RAS signalling (‘type I lesions’) were present in 7 out of 12 WGS cases, with an additional three cases having either type I or type II lesions, indicating that these events are central to the pathogenesis of ETP ALL. Consistent with this, pathway analysis incorporating both sequence and structural mutations demonstrated enrichment for lesions in these pathways in the 12 WGS cases (Supplementary Table 23). Across the entire cohort, 52 out of 64 (81.3%) ETP cases harboured mutations in these pathways, compared to 13 out of 42 (31%) non-ETP T-ALL cases (P < 0.0001; Fig. 3b and Supplementary Table 20). Forty-eight per cent of ETP cases had mutations in the PRC2 genes sequenced, SETD2 and EP300, compared to 12% of non-ETP ALL (P = 0.0001). This is probably an underestimate of the frequency of mutations perturbing chromatin modification, as not all PRC2 and histone-modifying genes have been sequenced. Pathway analysis of the gene expression profile of ETP ALL (Supplementary Table 24 and Supplementary Figs 23–25) demonstrated significant positive enrichment for genes mediating JAK-STAT signalling, and negative enrichment for T-cell receptor signalling genes in ETP ALL. In addition, flow cytometric intracellular phosphosignalling analysis of primary leukaemic cells demonstrated activation of RAS and JAK-STAT signalling pathways in ETP ALL cases (Supplementary Fig. 26). Furthermore, reconstruction of the transcriptional network of ETP ALL using ARACNE37 identified 30 gene networks (‘regulons’) with RUNX1 and IKZF1 observed to be hub genes of several of these regulons. Thus, alterations of these haematopoietic transcription factors are key determinants of the transcriptional profile of ETP ALL (Supplementary Table 25).
ETP ALL is a stem-cell leukaemia
The immunophenotype and gene expression of ETP ALL are similar to the murine early T-cell precursor3. However, detailed comparison of the gene expression profiles of ETP ALL and normal human haematopoietic progenitors has not been performed. Comparison of the gene expression profile of ETP ALL with those of purified normal38,39 and myeloid leukaemic40 haematopoietic stem cell and progenitor cell populations demonstrated marked negative enrichment of the gene expression profile of normal human early T-cell precursors (Supplementary Fig. 27). In contrast, the ETP ALL signature showed significant positive enrichment of the gene expression profile of normal human haematopoietic stem cells and granulocyte macrophage precursors. In addition, the ETP ALL signature demonstrated enrichment for a leukaemic stem-cell signature associated with poor outcome in acute myeloid leukaemia40, and a signature of poor outcome in IKZF1-mutated high-risk B-progenitor ALL41. Together, these data are compatible with the notion that the genetic alterations identified here result in gross maturational arrest and an aggressive poorly differentiated stem-cell-like leukaemia.
Discussion
Although the striking uniformity of clinical features, immunophenotype and transcriptional profile suggests a common underlying genetic alteration in ETP ALL, we identified a remarkable diversity of novel recurrent genetic alterations. Despite this diversity, the prevalence of mutations in genes involving cytokine receptor and RAS signalling, haematopoietic development and histone modification suggests a common pathogenesis for the establishment of the ETP leukaemic clone. Mutations known or predicted to result in activated cytokine receptor and RAS signalling are present in two-thirds of ETP cases, but only 19% of non-ETP T-ALL. This includes mutations in genes with known roles in leukaemogenesis as well as novel targets of mutation (JAK3, IL7R, IFNR1 and BRAF). The ability of the identified IL7R activating mutations to induce factor-independent growth of haematopoietic cells coupled with the known function of the other identified signalling mutations strongly supports a direct role for these alterations in leukaemic cell transformation. The high frequency of deleterious mutations in PRC2 genes suggests that disruption of PRC2-mediated gene silencing is a key event in the pathogenesis of this primitive leukaemia, but not more differentiated T-ALL cases. Several of the genes recurrently mutated in ETP are also mutated in inherited disorders (DNM2, EP300, GATA3, NRAS, KRAS, PHF6, RELN and RUNX1), and the mutational spectrum in several of these genes is similar between the inherited disorders and T-ALL. Thus, sequencing of additional T-ALL cases and other leukaemia genomes will be of great interest to fully examine the relationship of inherited and acquired lesions in leukaemogenesis.
Mutation of genes regulating cytokine receptor and/or RAS signalling pathway and epigenetic modification is a common feature of acute myeloid leukaemia but is less common in T- or B-lineage ALL (Supplementary Table 28)42. Although the gene expression profile of ETP ALL is similar to that of the murine ETP, it shows strong similarity to that of normal and myeloid leukaemic haematopoietic stem cells. This indicates that ETP ALL is distinct from non-ETP T-ALL, and in fact represents a neoplasm of a less mature haematopoietic progenitor or stem cell, with arrest at a very early maturational stage that retains the capacity for myeloid differentiation. This observation raises the possibility that treatment regimens used to treat acute myeloid leukaemia, such as those incorporating high dose cytarabine, and/or targeted therapies that inhibit cytokine receptor and JAK signalling may be beneficial in ETP ALL.
Methods Summary
Whole-genome sequencing was performed for tumour and normal DNA from 12 children with ETP ALL treated at St Jude Children’s Research Hospital. All cases fulfilled pathologic and immunophenotypic criteria for ETP ALL2. Tumour samples were obtained from diagnostic bone marrow aspirates or peripheral blood, and comprised at least 90% tumour cells. Matched non-tumour samples were obtained from remission blood or bone marrow aspirates with less than 1% leukaemic cells. Recurrence testing was performed using a cohort of 94 childhood T-ALL cases, comprising 52 ETP ALL cases from St Jude, the Children’s Oncology Group and the Associazione Italiana Ematologia de Oncologia Pediatrica (AIEOP), and 42 non-ETP T-ALL cases from St Jude. Whole-genome DNA sequencing was performed using a paired-end sequencing strategy as described in detail in the Supplementary Information. The frequency of the identified mutations in the recurrence cohort was determined using PCR amplification and Sanger sequencing and analysis of single-nucleotide polymorphism microarray data. The study was approved by the Institutional Review Boards of St Jude Children’s Research Hospital and Washington University.
Accession codes
Primary accessions
GenBank/EMBL/DDBJ
Gene Expression Omnibus
Data deposits
The sequence data and single nucleotide polymorphism microarray data have been deposited in the dbGaP database (http://www.ncbi.nlm.nih.gov/gap) under the accession number phs000340.v1.p1. Affymetrix U133A gene expression data have been deposited in the NCBI gene expression omnibus under GSE33315, and Affymetrix U133 Plus 2.0 PM gene expression data under accession GSE28703. The nucleotide sequence for the full-length ETV-INO80D transcript has been deposited in GenBank under accession JF736506. A public data portal for results from the St Jude – Washington University Pediatric Cancer Genome Project is available at http://www.explore.pediatriccancergenomeproject.org/.
References
Pui, C. H., Robison, L. L. & Look, A. T. Acute lymphoblastic leukaemia. Lancet 371, 1030–1043 (2008)
Coustan-Smith, E. et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 10, 147–156 (2009)
Rothenberg, E. V., Moore, J. E. & Yui, M. A. Launching the T-cell-lineage developmental programme. Nature Rev. Immunol. 8, 9–21 (2008)
Wada, H. et al. Adult T-cell progenitors retain myeloid potential. Nature 452, 768–772 (2008)
Wang, J. et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nature Methods 8, 652–654 (2011)
Stephens, P. J. et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40 (2011)
Bohlander, S. K. ETV6: a versatile player in leukemogenesis. Semin. Cancer Biol. 15, 162–174 (2005)
Shurtleff, S. A. et al. TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia 9, 1985–1989 (1995)
Barjesteh van Waalwijk van Doorn-Khosrovani, S. et al. Somatic heterozygous mutations in ETV6 (TEL) and frequent absence of ETV6 protein in acute myeloid leukemia. Oncogene 24, 4129–4137 (2005)
Yokota, S. et al. Mutational analysis of the N-ras gene in acute lymphoblastic leukemia: a study of 125 Japanese pediatric cases. Int. J. Hematol. 67, 379–387 (1998)
Kawamura, M. et al. Alterations of the p53, p21, p16, p15 and Ras genes in childhood T-cell acute lymphoblastic leukemia. Leuk. Res. 23, 115–126 (1999)
Flex, E. et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J. Exp. Med. 205, 751–758 (2008)
Weng, A. P. et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 (2004)
Paietta, E. et al. Activating FLT3 mutations in CD117/KIT+ T-cell acute lymphoblastic leukemias. Blood 104, 558–560 (2004)
Van Vlierberghe, P. et al. Activating FLT3 mutations in CD4+/CD8- pediatric T-cell acute lymphoblastic leukemias. Blood 106, 4414–4415 (2005)
Neumann, M. et al. High rate of FLT3 mutations in adult ETP-ALL. ASH Annu. Meet. Abstr. 116, 1031 (2010)
Van Vlierberghe, P. et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nature Genet. 42, 338–342 (2010)
Tosello, V. et al. WT1 mutations in T-ALL. Blood 114, 1038–1045 (2009)
Yamashita, Y. et al. Array-based genomic resequencing of human leukemia. Oncogene 29, 3723–3731 (2010)
Malinge, S. et al. Activating mutations in human acute megakaryoblastic leukemia. Blood 112, 4220–4226 (2008)
Ziegler, S. F. & Liu, Y. J. Thymic stromal lymphopoietin in normal and pathogenic T cell development and function. Nature Immunol. 7, 709–714 (2006)
Russell, L. J. et al. Deregulated expression of cytokine receptor gene, CRLF2, is involved in lymphoid transformation in B-cell precursor acute lymphoblastic leukemia. Blood 114, 2688–2698 (2009)
Mullighan, C. G. et al. Rearrangement of CRLF2 in B-progenitor- and Down syndrome-associated acute lymphoblastic leukemia. Nature Genet. 41, 1243–1246 (2009)
Shochat, C. et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J. Exp. Med. 208, 901–908 (2011)
Kleppe, M., Mentens, N., Tousseyn, T., Wlodarska, I. & Cools, J. MOHITO, a novel mouse cytokine-dependent T-cell line, enables studies of oncogenic signaling in the T-cell context. Haematologica 96, 779–783 (2011)
Hosoya, T., Maillard, I. & Engel, J. D. From the cradle to the grave: activities of GATA-3 throughout T-cell development and differentiation. Immunol. Rev. 238, 110–125 (2010)
Zahirieh, A. et al. Functional analysis of a novel GATA3 mutation in a family with the hypoparathyroidism, deafness, and renal dysplasia syndrome. J. Clin. Endocrinol. Metab. 90, 2445–2450 (2005)
Okuda, T., van Deursen, J., Hiebert, S. W., Grosveld, G. & Downing, J. R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 84, 321–330 (1996)
Downing, J. R. et al. An AML1/ETO fusion transcript is consistently detected by RNA-based polymerase chain reaction in acute myelogenous leukemia containing the (8;21)(q22;q22) translocation. Blood 81, 2860–2865 (1993)
Dicker, F. et al. Mutation analysis for RUNX1, MLL-PTD, FLT3-ITD, NPM1 and NRAS in 269 patients with MDS or secondary AML. Leukemia 24, 1528–1532 (2010)
Gaidzik, V. I. et al. RUNX1 mutations in acute myeloid leukemia: results from a comprehensive genetic and clinical analysis from the AML study group. J. Clin. Oncol. 29, 1364–1372 (2011)
Gelsi-Boyer, V. et al. Genome profiling of chronic myelomonocytic leukemia: frequent alterations of RAS and RUNX1 genes. BMC Cancer 8, 299 (2008)
Margueron, R. & Reinberg, D. The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349 (2011)
Morin, R. D. et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nature Genet. 42, 181–185 (2010)
Durieux, A. C., Prudhon, B., Guicheney, P. & Bitoun, M. Dynamin 2 and human diseases. J. Mol. Med. 88, 339–350 (2010)
Hong, S. E. et al. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nature Genet. 26, 93–96 (2000)
Margolin, A. A. et al. Reverse engineering cellular networks. Nature Protocols 1, 662–671 (2006)
Novershtern, N. et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144, 296–309 (2011)
Notta, F. et al. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 333, 218–221 (2011)
Eppert, K. et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nature Med. 17, 1086–1093 (2011)
Mullighan, C. G. et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N. Engl. J. Med. 360, 470–480 (2009)
Gilliland, D. G. Molecular genetics of human leukemias: new insights into therapy. Semin. Hematol. 39, 6–11 (2002)
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009)
Acknowledgements
We thank the many members of St Jude Children’s Research Hospital and The Genome Institute and Siteman Cancer Center at Washington University in St Louis for support. We thank H. Mulder for project sample management, M. Stine for assistance with data deposition, B. Pappas and S. Malone for information technology infrastructure, J. Morris, E. Walker, A. Merriman and G. Neale for performing single-nucleotide polymorphism and gene expression microarrays, W. Yang for assistance with analysis of genomic data, and J. Stokes for artwork. We thank the Tissue Resources Laboratory, the Flow Cytometry and Cell Sorting Core, and the Clinical Applications of Core Technology Laboratories of the Hartwell Center for Bioinformatics and Biotechnology of St Jude Children’s Research Hospital. We thank S. Kehoe of Beckman Coulter Genomics for assistance with Sanger sequencing. This work was funded by The St Jude Children’s Research Hospital – Washington University Pediatric Cancer Genome Project, ALSAC of St Jude Children’s Research Hospital, Cancer Center support grant P30 CA021765, NIH U01 GM 92666—PAAR4Kids, grants to R.K.W. from Washington University in St Louis and the National Human Genome Research Institute (NHGRI U54 HG003079), grants to the Children’s Oncology Group (NCI CA98543, CA98413, CA114766), and grants from Alex’s Lemonade Stand and St. Baldrick’s Foundation (to M.L.H.). S.L.H. was supported by a Haematology Society of Australasia and New Zealand New Investigator Scholarship. S.P.H. is the Ergen Family Chair in Pediatric Cancer. C.G.M. is a Pew Scholar in the Biomedical Sciences and a St. Baldrick’s Scholar.
Author information
Authors and Affiliations
Contributions
C.G.M., J.R.D., T.J.L., E.R.M. and R.K.W. designed the experiments. J.Z., L.D. and C.L. led data analysis. C.G.M., L.H., S.L.H., D.P.-T., J.R.C.-U., prepared patient samples and performed laboratory assays. R.S.F. and L.L.F. supervised whole genome sequencing data generation. K.C.B. managed data transfer. D.J.D. supervised the automated analysis pipeline. C.W.N. supervised computing infrastructure. J.E. performed transcriptome sequencing. J.R.C.-U., M.K. and J.C. performed IL7R mutation assays. K.A.S., M.L.H. and K.G.R performed phosphosignalling analyses. K.G.R. performed colony assays. X.C., M.R., J.W., G.W., J.B., D.M., J.M., D.Z., L.W., X.H., K.J.J. and C.C.H. performed sequence analysis. L.H., C.G.M. and J.M. analysed single-nucleotide polymorphism array data. M.P. performed telomere analysis. S.C.R. reviewed cytogenetic data. P.G. prepared Circos plots. D.A. and S.E. managed data. S.-C.C., J.M. and G.S. analysed gene expression microarray data. S.D., K.E., E.L., F.N. and J.E.D. collected and analysed gene expression data from normal and leukaemic stem and progenitor cells. S.B.P. developed the GRIN model and performed genomic pathway analyses. D.P. and C.C. performed association analyses between clinical and genetic variables and outcome. R.H. and R.W.K. performed EZH2 structural modelling. S.-C.C. performed pathway analysis. A.U. performed ARACNE analyses. D.C., E.C.-S., G.B., S.P.H., M.L.L., M.D., W.E.E., B.W., S.W., K.P.D., and C.-H.P. provided clinical samples and data. C.G.M., J.R.D and J.Z. wrote the manuscript. J.Z., L.D., L.H., G.W., S.L.H., D.P.-T., J.E., X.C., J.W., M.R., C.L., S.-C.C., L.W., J.M., K.G.R., S.B.P., A.U., J.B., P.G., R.H., R.W.K., M.P., D.J.M., D.Z., D.A., S.E., K.C.B., G.S., S.R., M.I.B., S.A.S., K.P.D., R.S.F., L.L.F., X.H., C.C.H., D.J.D., K.O., K.I.J., J.C.O., W.E.E., C.-H.P., C.W.N., T.J.L., E.R.M., R.K.W., J.R.D. and C.G.M. are part of the St Jude Children’s Research Hospital – Washington University Pediatric Cancer Genome Project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
The file contains Supplementary Results, Supplementary Tables 1, 5-10, 12, 16, 19-21,and 26-28, Supplementary Figures 1-27 with legends and additional references. (Please see contents page for full details). (PDF 6327 kb)
Supplementary Methods
The file contains Supplementary Methods and References. (PDF 275 kb)
Supplementary Tables
The file contains Supplementary Tables 2-4, 11, 13-15, 17-18 and 22-25. Please see Supplementary Information file for full descriptions. (ZIP 5376 kb)
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution-Non-Commercial-Share Alike licence (http://creativecommons.org/licenses/by-nc-sa/3.0/), which permits distribution, and reproduction in any medium, provided the original author and source are credited. This licence does not permit commercial exploitation, and derivative works must be licensed under the same or similar licence.
About this article
Cite this article
Zhang, J., Ding, L., Holmfeldt, L. et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012). https://doi.org/10.1038/nature10725
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nature10725
This article is cited by
-
Therapeutic challenges in peripheral T-cell lymphoma
Molecular Cancer (2024)
-
JAK/STAT in leukemia: a clinical update
Molecular Cancer (2024)
-
Characterisation of FLT3 alterations in childhood acute lymphoblastic leukaemia
British Journal of Cancer (2024)
-
A new genomic framework to categorize pediatric acute myeloid leukemia
Nature Genetics (2024)
-
Clinicohistoradiological and surgical outcome in diffuse midline glioma
Child's Nervous System (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
