
Abstract
In the latest 2016 World Health Organization classification of hematological malignancies, T-cell lymphoblastic lymphoma (T-LBL) and lymphoblastic leukemia (T-ALL) are grouped together into one entity called T-cell lymphoblastic leukemia/lymphoma (T-LBLL). However, the question of whether these entities represent one or two diseases remains. Multiple studies on driver alterations in T-ALL have led to a better understanding of the disease while, so far, little data on genetic profiles in T-LBL is available. We sought to define recurrent genetic alterations in T-LBL and provide a comprehensive comparison with T-ALL. Targeted whole-exome next-generation sequencing of 105 genes, multiplex ligation-dependent probe amplification, and quantitative PCR allowed comprehensive genotype assessment in 818, consecutive, unselected, newly diagnosed patients (342 T-LBL vs. 476 T-ALL). The median age at diagnosis was similar in T-LBL and T-ALL (17 vs. 15 years old, respectively; p = 0.2). Although we found commonly altered signaling pathways and co-occurring mutations, we identified recurrent dissimilarities in actionable gene alterations in T-LBL as compared to T-ALL. HOX abnormalities (TLX1 and TLX3 overexpression) were more frequent in T-ALL (5% of T-LBL vs 13% of T-ALL had TLX1 overexpression; p = 0.04 and 6% of T-LBL vs 17% of T-ALL had TLX3 overexpression; p = 0.006). The PI3K signaling pathway was significantly more frequently altered in T-LBL as compared to T-ALL (33% vs 19%; p < 0.001), especially through PIK3CA alterations (9% vs 2%; p < 0.001) with PIK3CAH1047 as the most common hotspot. Similarly, T-LBL genotypes were significantly enriched in alterations in genes coding for the EZH2 epigenetic regulator and in TP53 mutations (respectively, 13% vs 8%; p = 0.016 and 7% vs 2%; p < 0.001). This genetic landscape of T-LBLL identifies differential involvement of recurrent alterations in T-LBL as compared to T-ALL, thus contributing to better understanding and management of this rare disease.
Similar content being viewed by others


Introduction
Precursor T-cell malignancies are rare clonal hematopoietic stem cell neoplasms of lymphoid precursors that are committed to the T-cell lineage and mainly affect pediatric patients. By convention, the designation of T-cell lymphoblastic lymphoma (T-LBL) is used when the neoplasm is confined to a tissue lesion without or with only minimal blood or bone marrow involvement while T-cell lymphoblastic leukemia (T-ALL) is used when there is extensive blood involvement and/or >25% bone marrow infiltration. The World Health Organization denominated both T-ALL and T-LBL as T-lymphoblastic leukemia/lymphoma (T-LBLL) in the 2016 Revised World Health Organization classification of hematological malignancies but without further specification1. Despite this, similarities and differences in T-LBL and T-ALL regarding clinical course, phenotypic and molecular features have raised the question of whether these entities represent one disease or reflect two different diseases2. Molecular aspects of T-ALL have been widely explored and, although T-ALL and T-LBL share several common aberrations, clinical and multiomic strategies suggested that the two entities may have independent pathogenic requirements and dependencies3. Evidence that leukemic conversion originating from the T-LBL cell in lymphoid tissue can occur has been reported4,5. About 40% of relapsed T-LBL patients have bone marrow (BM) involvement, whereas less than 20% of T-LBL patients have histological evidence of BM involvement at diagnosis5. Conversely, about 20% of ALL patients undergo isolated extramedullary relapse (mainly central nervous system or testis) that could be considered lymphoma6. Gene expression analysis of T-LBL and T-ALL patient samples also showed different signatures for T-LBL and T-ALL in both children and adults, implying specific requirements to invade lymphoid tissues (T-LBL) or involve systemic compartments (T-ALL)7,8. Genes involved in angiogenesis, chemotactic response, and nodal metastases were more highly expressed in T-LBL7. Genes coding for proteins involved in cell-cell adhesion such as BCL2, S1P1, and ICAM1 have also been shown to be differentially expressed in T-LBL, leading to a blockade of tumor cell intravasation to blood9. This may explain why T-LBL and stromal cells are embedded in close proximity to lymphoid tissues. Phenotypically, it has been reported that T-LBL more frequently involves mature thymic cells than T-ALL, with a less frequent expression of myeloid antigens10,11,12. Finally, one DNA methylation study identified an epigenetic signature of differentially methylated CpG sites that segregates T-LBL from T-ALL. Those sites were associated with increased expression of membrane-associated protein domains13.
Understanding the significance of gene mutations in the diagnostic or prognosis of T-LBLL has already helped risk stratification and the development of individualized treatment14. T-ALL and T-LBL, however, remain highly aggressive malignant tumors, notably in adults. Despite event-free survival (EFS) rates of up to 50–90%, the overall survival rate after relapse is only ~3–27%15,16,17,18,19,20. A precise description of the genetic landscape of T-LBLL and specifically T-LBL is therefore desirable, in order to identify potential therapeutic targets and improve survival rates while reducing acute and long-term toxicities.
In this study, we analyzed the genetic landscape of 818 T-LBLL and identified significant differences in the incidence of PI3K/Akt, EZH2, and TP53 gene alterations between T-LBL and T-ALL. These alterations affect specific signaling pathways and may confer a susceptibility to recently developed targeted therapies.
Materials and methods
Patients
Diagnostic peripheral blood, bone marrow, or lymphoid tissue samples from 818 unselected adults and children with T-LBLL (476 patients with T-ALL and 342 with T-LBL), newly diagnosed between 1999 and 2020, were analyzed centrally in Necker-Enfants Malades Hospital (AP-HP, Paris France) after informed consent was obtained at diagnosis according to the Declaration of Helsinki. According to WHO criteria, if a patient presents with a mass lesion and lymphoblasts in the marrow, a value of >25% marrow blasts is used to define leukemia versus lymphoma1.
Gene mutation screening
A custom capture Nextera XT gene panel (Illumina, San Diego, CA) targeting all coding exons and their adjacent splice junctions of 105 genes was designed, based on available evidence in hematological neoplasms (Supplementary Table 2). DNA Libraries were prepared using Nextera Rapid Capture Enrichment protocol and underwent 2 × 150 bp paired-end sequencing on Illumina MiSeq sequencing system with MiSeq Reagent Kit v2 (Illumina). Briefly, sequence reads were filtered and mapped to the human genome (GRCh37/hg19) using in-house software (Polyweb, Institut Imagine, Paris). Annotated variants were selected after filtering out calls according to the following criteria: (1) coverage < 30×, <10 alternative reads or variant allelic fraction (VAF) < 7%; (2) polymorphisms described in dbSNP, 1000Genomes, EVS, Gnomad and EXAC with a calculated mean population frequency > 0.1%; (3) mutations with a frequency <2% in both T-LBL and T-ALL groups. Non-filtered variants were annotated using the somatic database COSMIC (version 78) and ProteinPaint (St Jude Children’s Research Hospital—Pediatric Cancer data portal). Lollipop plots were generated with ProteinPaint (https://pecan.stjude.org/#/ proteinpaint) and splice mutations were not depicted.
Molecular characterization of oncogenic drivers in T‑LBLL samples
Peripheral blood, bone marrow T-ALL samples, and lymphoid tissue T-LBL samples when available were analyzed for fusion transcripts (SIL-TAL1, CALM-AF10/PICALM-MLLT10), oncogenic transcripts (HOXA9, TLX1, and TLX3) and NOTCH1/FBXW7/RAS/PTEN mutations, as previously described21,22.
Multiplex ligation-dependent probe amplification (MLPA) analysis
MLPA analysis was performed using the MRC Holland (Amsterdam, The Netherlands) SALSA MLPA probe mix P383-A1 TALL according to the manufacturer’s recommendations. Polymerase chain reaction products were separated by capillary electrophoresis on an ABI-3130 device. Coffalyser software, available at http://www.mlpa.com, was used for the analysis.
Statistics
Comparisons for categorical variables between T-ALL and T-LBL subgroups were performed with Fisher’s exact test or Wilcoxon rank-sum test. Statistical analyses were performed with STATA software (STATA 12.0 Corporation, College Station, TX, USA) and StatAid R package23. All p-values were two-sided, with p < 0.05 denoting statistical significance. Circos plots and oncoplots were generated using R software. Strong correlations are indicated by large ellipses, whereas weak correlations are indicated by small ellipses. Co-occurrences and mutual exclusions in T-LBL and T-ALL patients were computed with the DISCOVER algorithm (version 0.9.3).
Results
Clinico-genomic comparison between T-ALL and T-LBL shows common features
Eight hundred and eighteen adults and children with T-LBLL (342 with T-LBL and 476 patients with T-ALL) were included in the study. Among the 476 T-ALL analyzed, 215 were adult patients (≥19 years old) and 261 were pediatric patients (<19 years old). Among the 342 T-LBL analyzed, 156 were adult patients (≥19 years old) and 186 were pediatric patients (<19 years old) (Supplementary Table 1). The median age at diagnosis was similar in T-LBL and T-ALL (17[1–72] vs 15[1–59] years old, respectively, (p = 0.2). 74% (249/335) of T-LBL vs 75% (357/476) of T-ALL were male (p > 0.9). CNS involvement occurred in 6% of T-LBL vs 11% of T-ALL (p = 0.11). Regarding oncogenetic classification, CALM-AF10 (PICALM-MLLT10) rearrangements were found in 4% of T-LBL vs 3% of T-ALL (p = 0.8). SIL-TAL1 rearrangements were also comparable in both groups (13% of T-LBL vs 14% of T-ALL; p > 0.9). Clinico-biological features of the cohort are summarized in Table 1.
Of the 818 T-LBLL samples, 804 harbored at least one pathogenic mutation or MLPA alteration (330/96.5% T-LBL and 474/99.6% T-ALL). The global representation of the mutation landscape in T-LBL and T-ALL is displayed as an oncoplot (Fig. 1A). Circos plots depicting co-occurring mutations in T-LBL and T-ALL are shown in Fig. 1B.

A Oncoplot of T-ALL vs T-LBL. B Circos plots depicting co-occurring alterations in T-LBL (left) and T-ALL (right).
Most alterations in T-LBLL affected NOTCH1/FBXW7 pathway genes (68% of cases), which was the most frequently involved pathway in both T-LBL and T-ALL. The second most frequently altered pathway was the cell cycle, with CDKN2A being deleted in 50% of T-LBL vs. 70% of T-ALL. Epigenetic regulating factors were commonly altered in both categories (53% overall). Other signaling pathways, including PI3K, JAK/STAT, and RAS (including KRAS, NRAS, NF1, and PTPN11), were also commonly altered in T-LBL and T-ALL. Genes coding for transcription factors were similarly mutated in T-LBL (40% of cases) and in T-ALL (44% of cases); p = 0.277 (Fig. 1A and supplementary Table 3).
Co-occurring gene alterations were also comparable between T-LBL and T-ALL (Fig. 1B). NOTCH1 mutations frequently co-occurred with CDKN2A deletions, FBXW7, PHF6, and BCL11B mutations and are significantly less associated with PTEN alterations.
A thorough, comprehensive genetic landscape analysis of T-LBL, however, identified recurrent dissimilarities in oncogene alterations as compared to T-ALL
Molecular dissimilarities in T-LBL vs T-ALL
Several oncogenes were significantly differentially distributed and are described below. Gene and pathway alterations in T-LBL and T-LBL are depicted and detailed in Fig. 2 and supplementary Table 3. Regarding oncogenic drivers, NKL homeotic abnormalities (TLX1 and TLX3) were more frequent in T-ALL as compared to T-LBL. TLX1 overexpression was found in 6% of T-LBL vs. 13% of T-ALL (p = 0.04) and TLX3 overexpression in 7% of T-LBL vs. 17% of T-ALL (p = 0.006).

The significance level of co-occurrences or mutual exclusions are depicted as followed: * indicating a p-value < 0.05, ** a p-value < 0.01, and *** a p-value < 0.001.
NOTCH1/FBXW7 pathway
NOTCH1 alterations were identified in 52% (170/330) of T-LBL patients versus 72% (342/474) of T-ALL (p < 0.001). Mutations mainly clustered in the NOTCH1 HD domain (56% of NOTCH1 mutations in T-LBL vs 42% of T-ALL; p = 0.086) (Supplementary Table 4). FBXW7 mutations were found in 24% of T-LBL vs 20% of T-ALL (p = 0.226). However, the co-occurrence mutational profile of FBXW7 differs between T-LBL and T-ALL. FBXW7 mutations were significantly more commonly associated with PTEN and STAT5B alterations in T-LBL while FBXW7 mutations were more commonly associated with JAK1 and RUNX1 mutations in T-ALL (Fig. 3).

Corrplot showing the co-mutations in: A T-LBL patients and B T-ALL patients. The colors of the scale bar denote the nature of the correlation, with +1 indicating a perfectly positive correlation (blue) and −1 indicating a perfectly negative correlation (red) between two alterations. The significance level of co-occurrences or mutual exclusions are depicted as followed: * indicating a p-value < 0.05, ** a p-value < 0.01, and *** a p-value < 0.001.
Cell cycle
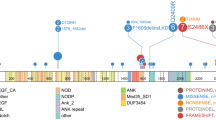
We observed significantly fewer CDKN2A alterations in T-LBL (50% of T-LBL vs 70% of T-ALL; p < 0.001). Conversely, T-LBL were enriched in TP53 mutations (7% of T-LBL vs 2% of T-ALL; p < 0.001). TP53 mutations had no significant co-occurrence with other gene alterations in T-ALL whereas they were less significantly associated with CDKN2A deletions in T-ALL (Fig. 3). TP53 mutations mainly affected the p53 DNA-binding domain of the protein (exon 5–8) in both T-LBL and T-ALL (Fig. 4A). No differential incidence of mutations between adult and pediatric T-LBL regarding TP53 was found (Supplementary Table 5).

Lollipop plots indicating the observed mutations and their consequences for: A TP53. B EZH2. C PIK3CA. D PIK3R1. E AKT1.
Epigenetic deregulation
Epigenetic regulating factors were more frequently altered in T-ALL (52% of T-LBL vs 60% of T-ALL cases; p = 0.02). PHF6 was the most frequently altered gene in this category, with mutations detected in 29% (232/804) of patients (71/330, 22% of T-LBL vs 161/474, 34% of T-ALL; p < 0.001). T-ALL patients were significantly enriched in CTCF and EED mutations as compared to T-LBL (respectively, 1% of T-LBL vs 5% of T-ALL; p = 0.001 and 1% of T-LBL vs 4% of T-ALL; p = 0.002). In addition, T-LBL patients were enriched in EZH2 alterations (43/330, 13% of T-LBL vs 36/474, 8% of T-ALL; p = 0.016).
The main alterations were deletions (28% of EZH2 alteration in T-LBL vs 31% in T-ALL) and missense mutations (42% in T-LBL vs 25% in T-ALL) (Supplementary Table 4). Of note, EZH2 alterations were significantly less commonly associated with NOTCH1 and FBXW7 mutations in T-LBL (Fig. 3A). Mutations were mainly located in exons 16 to 20 in T-ALL and in T-LBL, affecting the SET domain of the protein (Fig. 4B).
Finally, KMT2D was mutated in 14% of T-LBL vs 8% of T-ALL (p = 0.001). The distribution of KMT2D variants in adult and pediatric patients is pictured in Supplementary Fig. 1. No differential incidence of alterations between adult and pediatric T-LBL regarding EZH2 or KMT2D was found (Supplementary Table 5).
JAK/STAT signaling pathways
The JAK/STAT signaling pathway was significantly more affected in T-ALL vs T-LBL (207/474, 43% vs 110/330, 33%, respectively; p = 0.004), partly due to the higher incidence of DNM2 mutations in T-ALL (70/474, 15% of T-ALL vs 23/330, 7% of T-LBL; p < 0.001). Regarding other major genes involved in JAK/STAT signaling, IL7R, JAK1, JAK3, and STAT5B were mutated in 10%, 6%, 14%, and 6% in T-LBLL, respectively, with no incidence difference between T-LBL and T-ALL (Fig. 1A and Supplementary Table 3).
PI3K signaling pathway
Evaluation of PI3K signaling pathway gene mutations in T-LBL highlights important dissimilarities with T-ALL. Overall, 5% (38/804) of patients had PIK3CA mutations (30/330, with 9% in T-LBL vs 8/474, 2% in T-ALL; p < 0.001), comprising 39 mutations (31 in T-LBL and 8 in T-ALL). Of these, 38 were missense mutations and one was a nonsense mutation. Interestingly, the most frequent mutation of PIK3CA in T-LBL was missense mutation at H1047 in exon 21 (kinase domain) (11/31 mutations, 35%) while T-ALL featured scattered mutations without preferential location. PIK3CA mutations are depicted in Fig. 4C and detailed in Supplementary Table 6. While no significant co-occurrence between PIK3CA and other gene alterations was reported in T-ALL, PIK3CA mutations were significantly less associated with NOTCH1, PHF6, PTEN, and JAK3 alterations in T-LBL (Fig. 3).
PIK3R1 mutations were observed in 5% (42/804) of patients (24/330, 7% of T-LBL vs 18/474, 4% of T-ALL; p = 0.036) with 43 mutations reported (25 in T-LBL and 18 in T-ALL) featuring 25 missense, 11 indels, 3 frameshifts, and 4 splicing mutation. Most of the mutations observed were in the Inter-Src homology 2 (iSH2) helical domain of the protein, especially in exon 13. PIK3R1 mutations were significantly less associated with NOTCH1, PHF6, and PIK3CA alterations whilst no association or exclusion were observed in T-ALL. Details of PIK3R1 mutations (splicing mutations excluded) are depicted in Fig. 4D.
AKT1 were mutated in 1% (10/804) of patients overall, including 2% (8/330) of T-LBL vs 2/474, 0.4% of T-ALL; p = 0.019). 12 missense mutations were reported (8 mutations in T-LBL and 4 mutations in T-ALL). Eighty three percent (10/12) of mutations were E17K in exon 3 (kinase domain). Seven AKT1 E17K mutations were identified in T-LBL and 2 in T-ALL (Fig. 4E).
The incidence of PTEN alterations was comparable (52/330, 16% of T-LBL vs 69/474, 15% of T-ALL; p = 0.689) with PTEN deletions identified in 27% of PTEN altered T-LBL cases and in 22% of T-ALL cases (Supplementary Table 4).
No difference was seen in the incidence of mutations between adult and pediatric T-LBL regarding PIK3CA, PIK3R1, and AKT1. (Supplementary Table 5).
Discussion
This study provides a comprehensive genetic study of T-LBLL and demonstrates that the T-LBL oncogenetic landscape differs from T-ALL.
The incidence of specific driver oncogenic rearrangement was significantly different in T-LBL and T-ALL regarding HOX abnormalities, since T-ALL were relatively enriched in TLX1 and TLX3 overexpression. TLX1 overexpression, found in a “proliferative” molecular cluster, is associated with a better outcome in T-ALL24 while the prognostic impact of TLX3 overexpression, found in a “TLX” molecular cluster, remains contested25,26. So far, little is known about oncogenic drivers in T-LBL patients. Few T-LBL patients benefit from RT-qPCR or rearrangement screening, as is systematically performed for T-ALL.
T-LBL also demonstrated differences in recurrent gene alterations affecting actionable signaling pathways as compared to T-ALL.
Most importantly, NGS-based molecular screening reveals recurrent PI3K signaling pathway alterations in T-LBL as compared to T-ALL. Nine percent of T-LBL patients had PIK3CA mutations, mainly involving exons 10 and 21. This is slightly higher than previously reported in pediatric and adult T-LBL cohorts (7% and 6%, respectively),27,28. PIK3R1 mutations in our cohort were also higher compared to previous observations (7% vs 4%)27. Regarding T-ALL, there were few PIK3CA (2%), PIK3R1 (4%), and AKT1 (0.4%) mutations, which is consistent with the literature, although slightly lower than previously reported, albeit limited, cohorts29,30. A recent NGS-based study of 87 adult T-LBL did not identify significant PIK3CA, PIK3R1, or AKT1 mutations31. PIK3CA mutations were significantly less associated with NOTCH1, PHF6, and PTEN alteration in T-LBL, suggesting a specific oncogenic role. The PI3K/Akt signaling pathway plays an important role in the early stages of thymocyte development, specifically for the transition of double-negative to double-positive thymocytes. Whereas a loss of regular PI3K/Akt signaling leads to defective thymocyte development, its aberrant activation triggers uncontrolled cell survival, growth, and proliferation that may lead to the formation of T-cell lymphomas32. The PI3K/Akt pathway also has a crucial role in cell-to-cell adhesion and is implicated in tumor-induced extracellular matrix reorganization and tumor dissemination33,34,35,36. We hypothesize that activating mutations in this pathway might affect thymocyte-stroma interaction to promote tumor formation and dissemination preferentially in T-LBL. Clinically, specific inhibition of PIK3CA in PIK3CA-related overgrowth CLOVES syndrome patients induces spectacular disease regression37. Interestingly, the 19 T-LBL patients in this cohort showed a spectrum of mutations very similar to those reported in this study, with PIK3CAH1047 as the most frequent alteration (Fig. 4C). PI3K/Akt pathway alterations and specifically PIK3CA alterations are frequent targetable lesions in solid tumors. Promising clinical trials have led to FDA approval for alpelisib in metastatic breast cancer38. Our results show that mutations in the PI3K/Akt signaling pathway are recurrent features and potentially actionable targets in T-LBL.
Additional genomic differences between T-LBL and T-ALL affect NOTCH1, CDKN2A genes, and epigenetic regulating factors. NOTCH1 mutations affect the HD or TAD/PEST domain of the protein, resulting in increased NOTCH1 signaling. These mutations have been widely described in T-ALL, when they are found in up to 70% of diagnostic samples39,40. NOTCH pathway mutations in T-ALL have been associated with a favorable outcome and improved steroid responses41,42. NOTCH1 mutations have been reported in 55–60% of pediatric T-LBL2,28,43 and 36–52% of adult T-LBL31,44, while we now report NOTCH1 mutations in 52% of T-LBL patients. These mutations were also associated with a favorable prognostic effect31,43. Interestingly, T-ALL were significantly enriched in NOTCH1 mutations as compared to T-LBL, particularly those leading to single in-frame mutations, which were absent in T-LBL, compared to 17% of T-LBL.
With respect to epigenetic abnormalities, the EZH2 gene was more frequently mutated in T-LBL. In previous studies, EZH2 was mutated in 7% of T-ALL adult cases while EZH2 gain was observed in 13% of T-LBL pediatric cases with SNP array28,31. Loss-of-function EZH2 mutations were found in T-ALL, along with deletions and mutations of other Polycomb Repressive Complex 2 (PRC2) subunits45,46,47. EZH2 is the functional enzymatic component of PRC2 and is responsible for its methylation activity. PRC2 loss-of-function alterations can profoundly reshape the genetic and epigenetic landscape of T-ALL, leading to the reactivation of stem cell programs that cooperate with Bromodomain and Extraterminal (BET) proteins to sustain T-ALL. We recently identified a targetable vulnerability to BET inhibition in PRC2-altered T-ALL patients47. The present data suggest that EZH2-altered T-LBL patients should also be considered for such targeted therapy. On the other hand, other mutations in PRC2 subunits (i.e., SUZ12) and in epigenetic factor PHF6 were enriched in T-ALL. Similar to what has been reported, SUZ12 inactivation and PHF6 alterations significantly co-occurred with JAK3 mutations in our T-ALL patients as compared to T-LBL48,49. Based on our results, selective PRC2 subunits alterations seem to be preferentially associated with leukemic or tumoral involvement and should be further explored. In the same manner, PHF6 gene does not seem to play a major role in lymphomagenesis as compared to what has been reported in T-ALL50. In contrast to T-ALL cell lines, PHF6 mutations have been associated with a favorable outcome in adult T-LBL patients31. The role PHF6 plays in leukemogenesis is still actively under investigation and further study is therefore required to assess its true role and significance in T-LBLL patients.
Intriguingly, we found a higher incidence of KMT2D mutation, another epigenetic modifier, in T-LBL vs T-ALL. KMT2D have been previously shown to be associated with poor prognosis in pediatric T-LBL28. We reported a similar KMT2D mutation incidence (14% vs 13%) in T-LBL, but we did not observe a differential incidence of KMT2D mutation in pediatric vs. adult patients. In addition, we found that mutations in T-LBL were rather localized in a region ranging from 2000 to 3000 amino acids, as previously described, and, specifically to our cohort, in exon 11. For T-ALL, we found 8% of patients with KMT2D mutation which is higher than data previously reported, albeit in limited cohorts of purely pediatric patients (2% and 3%)51,52. Although KMT2D gene has been very little studied in T-LBLL, it seems to have an important role in leukemogenesis specifically in T-LBL. Because of a heterogenous distribution of mutations, further studies are needed for better characterization.
T-LBL were also highly enriched in TP53 missense mutations as compared to T-ALL. TP53 is the most commonly mutated somatic gene in human cancer and is often associated with a poor outcome in hematological malignancies53. The TP53 targeting drug APR 246 (eprenetapopt) has recently shown promising results in myeloid malignancies with mutant TP5354,55. APR 246 is a small molecule that restores wild-type p53 functions in TP53-mutant cells and could be part of the future of care in T-LBL/T-ALL harboring TP53 Mutation.
The JAK/STAT signaling pathway was frequently altered in T-LBLL (39% of cases), particularly in T-ALL, with JAK3 as the main mutated gene in both T-LBL (12%) and T-ALL (15%). The incidence of IL7R was frequent and similar in T-LBL and T-ALL (about 10%). IL7R/JAK/STAT pathway alterations are known to participate in leukemic development and are associated with reduced steroid sensitivity and poor clinical outcome56, although this may improve with targeted use of JAK inhibitors (tofacitinib and ruxolitinib)57,58,59.
In conclusion, we provide the largest comparative study exploring the oncogenic landscape of T-LBL vs T-ALL and reveal for the first time significant preferential alterations in T-LBL vs T-ALL (PI3K/Akt pathway, TP53, and PRC2/EZH2 alterations) using a pan-exon NGS-based approach. We identified recurrent mutations in targetable oncogenic signaling pathways in T-LBL and suggest that certain somatic abnormalities may affect the degree of tissue dissemination to blood and or bone marrow. Nevertheless, further prospective studies in T-LBL and T-ALL patients homogeneously treated are needed to better characterize the impact of genomic alterations in these rare diseases.
Data availability
Data will be made publicly available.
References
Arber, D. A., Orazi, A., Hasserjian, R., Thiele, J., Borowitz, M. J., le Beau, M. M. et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 127, 2391–2405 (2016).
Kroeze, E., Loeffen, J. L. C., Poort, V. M. & Meijerink, J. P. P. T-cell lymphoblastic lymphoma and leukemia: different diseases from a common premalignant progenitor? Blood Adv 4, 3466–3473 (2020).
van der Zwet, J. C. G., Cordo’, V., Canté-Barrett, K. & Meijerink, J. P. P. Multi-omic approaches to improve outcome for T-cell acute lymphoblastic leukemia patients. Adv Biol Regul 74, 100647 (2019).
Burkhardt, B., Zimmermann, M., Oschlies, I., Niggli, F., Mann, G., Parwaresch, R. et al. The impact of age and gender on biology, clinical features and treatment outcome of non-Hodgkin lymphoma in childhood and adolescence. Br J Haematol 131, 39–49 (2005).
Burkhardt, B., Reiter, A., Landmann, E., Lang, P., Lassay, L., Dickerhoff, R. et al. Poor outcome for children and adolescents with progressive disease or relapse of lymphoblastic lymphoma: a report from the Berlin-Frankfurt-Muenster group. J Clin Oncol 27, 3363–3369 (2009).
Hagedorn, N., Acquaviva, C., Fronkova, E., Von Stackelberg, A., Barth, A., Zur Stadt, U. et al. Submicroscopic bone marrow involvement in isolated extramedullary relapses in childhood acute lymphoblastic leukemia: a more precise definition of “isolated” and its possible clinical implications, a collaborative study of the Resistant Disease Committee of the International BFM study group. Blood 110, 4022–4029 (2007).
Basso, K., Mussolin, L., Lettieri, A., Brahmachary, M., Lim, W. K., Califano, A. et al. T-cell lymphoblastic lymphoma shows differences and similarities with T-cell acute lymphoblastic leukemia by genomic and gene expression analyses. Genes Chromosomes Cancer 50, 1063–1075 (2011).
Bonn, B. R., Huge, A., Rohde, M., Oschlies, I., Klapper, W., Voss, R. et al. Whole exome sequencing hints at a unique mutational profile of paediatric T-cell lymphoblastic lymphoma. Br J Haematol vol. 168 308–313 (2015).
Feng, H., Stachura, D. L., White, R. M., Gutierrez, A., Zhang, L., Sanda, T. et al. T-lymphoblastic lymphoma cells express high levels of BCL2, S1P1, and ICAM1, leading to a blockade of tumor cell intravasation. Cancer Cell 18, 353–366 (2010).
Patel, J. L., Smith, L. M., Anderson, J., Abromowitch, M., Campana, D., Jacobsen, J. et al. The immunophenotype of T-lymphoblastic lymphoma in children and adolescents: A children’s oncology group report. Br J Haematol 159, 454–461 (2012).
Bernard, A., Boumsell, L., Reinherz, E., Nadler, L., Ritz, J., Coppin, H. et al. Cell surface characterization of malignant T cells from lymphoblastic lymphoma using monoclonal antibodies: evidence for phenotypic differences between malignant T cells from patients with acute lymphoblastic leukemia and lymphoblastic lymphoma. Blood 57, 1105–1110 (1981).
Uyttebroeck, A., Vanhentenrijk, V., Hagemeijer, A., Boeckx, N., Renard, M., Wlodarska, I. et al. Is there a difference in childhood T-cell acute lymphoblastic leukaemia and T-cell lymphoblastic lymphoma? Leuk Lymphoma 48, 1745–1754 (2007).
Haider, Z., Landfors, M., Golovleva, I., Erlanson, M., Schmiegelow, K., Flægstad, T. et al. DNA methylation and copy number variation profiling of T-cell lymphoblastic leukemia and lymphoma. Blood Cancer J 10, 45 (2020).
Terwilliger, T. & Abdul-Hay, M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. 7, e577 (2017).
Pillon, M., Piglione, M., Garaventa, A., Conter, V., Giuliano, M., Arcamone, G. et al. Long-term results of AIEOP LNH-92 protocol for the treatment of pediatric lymphoblastic lymphoma: a report of the Italian association of pediatric hematology and oncology. Pediatr Blood Cancer 53, 953–959 (2009).
Goldberg, J. M., Silverman, L. B., Levy, D. E., Dalton, V. K., Gelber, R. D., Lehmann, L. et al. Childhood T-cell acute lymphoblastic leukemia: The Dana-Farber Cancer Institute Acute Lymphoblastic Leukemia Consortium experience. J Clin Oncol vol. 21 3616–3622 (2003).
Sandlund, J. T., Pui, C. H., Zhou, Y., Behm, F. G., Onciu, M., Razzouk, B. I. et al. Effective treatment of advanced-stage childhood lymphoblastic lymphoma without prophylactic cranial irradiation: Results of St Jude NHL13 study. Leukemia 23, 1127–1130 (2009).
Huguet, F., Chevret, S., Leguay, T., Thomas, X., Boissel, N., Escoffre-Barbe, M. et al. Intensified therapy of acute lymphoblastic leukemia in adults: Report of the randomized GRAALL-2005 clinical trial. J Clin Oncol 36, 2514–2523 (2018).
Oudot, C., Auclerc, M. F., Levy, V., Porcher, R., Piguet, C., Perel, Y. et al. Prognostic factors for leukemic induction failure in children with acute lymphoblastic leukemia and outcome after salvage therapy: The FRALLE 93 study. J Clin Oncol 26, 1496–1503 (2008).
Burkhardt, B., Taj, M., Garnier, N., Minard-Colin, V., Hazar, V., Mellgren, K. et al. Treatment and outcome analysis of 639 relapsed non-hodgkin lymphomas in children and adolescents and resulting treatment recommendations. Cancers 13, 2075 (2021).
Trinquand, A., Tanguy-Schmidt, A., Abdelali, R. Ben, Lambert, J., Beldjord, K., Lengliné, E. et al. Toward a NOTCH1/FBXW7/RAS/PTEN-based oncogenetic risk classification of adult T-Cell acute lymphoblastic leukemia: a group for research in adult acute lymphoblastic leukemia study. J Clin Oncol 31, 4333–4342 (2013).
Bond, J., Marchand, T., Touzart, A., Cieslak, A., Trinquand, A., Sutton, L. et al. An early thymic precursor phenotype predicts outcome exclusively in HOXA-overexpressing adult T-cell acute lymphoblastic leukemia: A group for research in adult acute lymphoblastic leukemia study. Haematologica 101, 732–740 (2016).
Alcazer, V. StatAid: An R package with a graphical user interface for data analysis. J. Open Source Softw 5, 2630 (2020).
Dadi, S., Le Noir, S., Payet-Bornet, D., Lhermitte, L., Zacarias-Cabeza, J., Bergeron, J. et al. TLX Homeodomain Oncogenes Mediate T Cell Maturation Arrest in T-ALL via Interaction with ETS1 and Suppression of TCRα Gene Expression. Cancer Cell 21, 563–576 (2012).
Cavé, H., Suciu, S., Preudhomme, C., Poppe, B., Robert, A., Uyttebroeck, A. et al. Clinical significance of HOX11L2 expression linked to t(5;14)(q35;q32), of HOX11 expression, and of SIL-TAL fusion in childhood T-cell malignancies: Results of EORTC studies 58881 and 58951. Blood 103, 442–450 (2004).
Ballerini, P., Landman-Parker, J., Cayuela, J. M., Asnafi, V., Labopin, M., Gandemer, V. et al. Impact of genotype on survival of children with T-cell acute lymphoblastic leukemia treated according to the French protocol FRALLE-93: the effect of TLX3/HOX11L2 gene expression on outcome. Haematologica 93, 1658–1665 (2008).
Balbach, S. T., Makarova, O., Bonn, B. R., Zimmermann, M., Oschlies, I., Klapper, W. et al. Proposal of a genetic classifier for risk group stratification in pediatric T-cell lymphoblastic lymphoma reveals differences from adult T-cell lymphoblastic leukemia. Leukemia vol. 30 970–973 (2016).
Khanam, T., Sandmann, S., Seggewiss, J., Ruether, C., Zimmermann, M., Norvil, A. B. et al. I Integrative genomic analysis of pediatric T-cell lymphoblastic lymphoma reveals candidates of clinical significance. Blood 137, 2347–2359 (2021).
Gutierrez, A., Sanda, T., Grebliunaite, R., Carracedo, A., Salmena, L., Ahn, Y. et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood 114, 647–650 (2009).
Zuurbier, L., Petricoin, E. F., Vuerhard, M. J., Calvert, V., Kooi, C., Buijs- Gladdines, J. G. C. A. M. et al. The significance of PTEN and AKT aberrations in pediatric T-cell acute lymphoblastic leukemia. Haematologica 97, 1405–1413 (2012).
Li, Z., Song, Y., Zhang, Y., Li, C., Wang, Y., Xue, W. et al. Genomic and outcome analysis of adult T-cell lymphoblastic lymphoma. Haematologica vol. 105 E107–E110 (2020).
Fayard, E., Moncayo, G., Hemmings, B. A. & Holländer, G. A. Phosphatidylinositol 3-kinase signaling in thymocytes: The need for stringent control. Sci Signal. 3, 1–13 (2010).
Chen, Y., Hou, Q., Yan, W., J, L., D, C., Z, L. et al. PIK3CA is critical for the proliferation, invasiveness, and drug resistance of human tongue carcinoma cells. Oncol Res. 19, 563–571 (2011).
Matsuoka, T., Yashiro, M., Nishioka, N., Hirakawa, K., Olden, K. & Roberts, J. D. PI3K/Akt signalling is required for the attachment and spreading, and growth in vivo of metastatic scirrhous gastric carcinoma. Br J Cancer 106, 1535–1542 (2012).
Li, B., Xu, W., Lam, A., Y, W., HF, H., XY, G. et al. S Significance of PI3K/AKT signaling pathway in metastasis of esophageal squamous cell carcinoma and its potential as a target for anti-metastasis therapy. Oncotarget 8, 38755–38766 (2017).
Hirsch, E., Ciraolo, E., Franco, I., Ghigo, A. & Martini, M. PI3K in cancer–stroma interactions: bad in seed and ugly in soil. Oncogene 2014 33:24 33, 3083–3090 (2013).
Venot, Q., Blanc, T., Rabia, S. H., Berteloot, L., Ladraa, S. & Duong, J. et al. Targeted therapy in patients with PIK3CA-related overgrowth syndrome. Nature 558, 540–546 (2021).
André, F., Ciruelos, E., Rubovszky, G., Campone, M., Loibl, S., Rugo, H. S. et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor–Positive Advanced Breast Cancer. N Engl J Med. 380, 1929–1940 (2019).
Weng, A. P., Ferrando, A. A., Lee, W., Morris IV, J. P., Silverman, L. B., Sanchez- Irizarry, C. et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306, 269–271 (2004).
Aref, S., el Agdar, M., Salama, O., Zeid, T. A. & Sabry, M. Significance of NOTCH1 mutations detections in T-acute lymphoblastic leukemia patients. Cancer Biomark 27, 157–162 (2020).
Clappier, E., Collette, S., Grardel, N., Girard, S., Suarez, L., Brunie, G. et al. NOTCH1 and FBXW7 mutations have a favorable impact on early response to treatment, but not on outcome, in children with T-cell acute lymphoblastic leukemia (T-ALL) treated on EORTC trials 58881 and 58951. Leukemia 24, 2023–2031 (2010).
Breit, S., Stanulla, M., Flohr, T., Schrappe, M., Ludwig, W. D., Tolle, G. et al. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood 108, 1151–1157 (2006).
Bonn, B. R., Rohde, M., Zimmermann, M., Krieger, D., Oschlies, I., Niggli, F. et al. Incidence and prognostic relevance of genetic variations in T-cell lymphoblastic lymphoma in childhood and adolescence. Blood 121, 3153–3160 (2013).
Lepretre, S., Touzart, A., Vermeulin, T., Picquenot, J. M., Tanguy-Schmidt, A., Salles, G. et al. Pediatric-like acute lymphoblastic leukemia therapy in adults with lymphoblastic lymphoma: the GRAALL-LYSA LL03 study. J Clin Oncol. 34, 572–580 (2016).
Schäfer, V., Ernst, J., Rinke, J., Winkelmann, N., Beck, J. F., Hochhaus, A. et al. EZH2 mutations and promoter hypermethylation in childhood acute lymphoblastic leukemia. J Cancer Res Clin Oncol 142, 1641–1650 (2016).
Ntziachristos, P., Tsirigos, A., Vlierberghe, P. Van, Nedjic, J., Trimarchi, T.,Flaherty, M. S. et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med 18, 296–301 (2012).
Andrieu, G. P., Kohn, M., Simonin, M., Smith, C., Cieslak, A., Dourthe, M.-E. et al. PRC2 loss of function confers a targetable vulnerability to BET proteins in T-ALL. Blood 138, 1855–1869 (2021).
Broux, M., Prieto, C., Demeyer, S., Bempt, M. vanden, Alberti-Servera, L., Lodewijckx, I. et al. Suz12 inactivation cooperates with JAK3 mutant signaling in the development of T-cell acute lymphoblastic leukemia. Blood 134, 1323-1336 (2019).
Yuan, S., Wang, X., Hou, S., Guo, T., Lan, Y., Yang, S. et al. PHF6 and JAK3 mutations cooperate to drive T-cell acute lymphoblastic leukemia progression. Leukemia 36, 370–382 (2022).
Kurzer, J. H. & Weinberg, O. K. PHF6 mutations in hematologic malignancies. Front Oncol 11, 704471 (2021).
Liu, Y., Easton, J., Shao, Y., Maciaszek, J., Wang, Z., Wilkinson, M. R. et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet 49, 1211–1218 (2017).
Zhang, H., Wang, H., Qian, X., Gao, S., Xia, J., Liu, J. et al Genetic mutational analysis of pediatric acute lymphoblastic leukemia from a single center in China using exon sequencing. BMC Cancer 20, 211 (2020).
Stengel, A., Kern, W., Haferlach, T., Schnittger, S., Zenger, M. & Haferlach, C. Comparison of TP53 Alterations in Hematological Malignancies. Blood 126, 4819–4819 (2015).
Cluzeau, T., Sebert, M., Rahmé, R., Cuzzubbo, S., Lehmann-Che, J., Madelaine, I. et al. Eprenetapopt Plus Azacitidine In TP53-mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase Ii Study by the Groupe Francophone des Myélodysplasies (GFM). J Clin Oncol 39, 1575–1583 (2021).
Sallman, D. A., DeZern, A. E., Garcia-Manero, G., Steensma, D. P., Roboz, G. J., Sekeres, M. A. et al. Eprenetapopt (APR-246) and Azacitidine in TP53 -Mutant Myelodysplastic Syndromes. J Clin Oncol 39, 1584–1594 (2021).
Li, Y., Buijs-Gladdines, J. G. C. A. M., Canté-Barrett, K., Stubbs, A. P., Vroegindeweij, E. M., Smits, W. K. et al. IL-7 Receptor Mutations and Steroid Resistance in Pediatric T Cell Acute Lymphoblastic Leukemia: A Genome Sequencing Study. PLoS Med 13, e1002200 (2016).
Delgado-Martin, C., Meyer, L. K., Huang, B. J., Shimano, K. A., Zinter, M. S., Nguyen, J. v et al. JAK/STAT pathway inhibition overcomes IL7-induced glucocorticoid resistance in a subset of human T-cell acute lymphoblastic leukemias. Leukemia 2017 31:12 31, 2568–2576 (2017).
Senkevitch, E., Hixon, J., Andrews, C., Barata, J. T., Li, W. & Durum, S. The JAK inhibitor ruxolitinib is effective in treating T cell acute lymphoblastic leukemia with gain of function mutations in IL-7R alpha. Blood 126, 1330–1330 (2015).
Cabannes, A., Schmidt, A., Brissot, E., Balsat, M., Maury, S., Isnard, F. et al. The combination of Venetoclax and Tofacitinib Induced Hematological Responses in Patients with Relapse/ Refractory T-ALL with BCL2 Expression and Surface IL7R Expression or IL7R-Pathway Mutations (On Behalf of the GRAALL). Blood 134, 1339 (2019).
Acknowledgements
We would like to thank all participants in the GRAALL-2003 and GRAALL-2005 study groups, the SFCE and the investigators of the 16 SFCE centers involved in collection and provision of data and patient samples, and V. Lheritier for collection of clinical data. The GRAALL was supported by grants P0200701 and P030425/AOM03081 from the Programme Hospitalier de Recherche Clinique, Ministère de l’Emploi et de la Solidarité, France and the Swiss Federal Government in Switzerland. Samples were collected and processed by the AP-HP “Direction de Recherche Clinique” Tumor Bank at Necker-Enfants Malades. M.S. was supported by « Action Leucémie », « Soutien pour la formation à la recherche translationnelle en cancérologie, Plan cancer 2014–2019 », and the “Ligue Nationale Contre le Cancer ». This work was supported by grants to Necker laboratory from the “Association Laurette Fugain”, Association pour la Recherche contre le Cancer (Equipe labellisée), Institut National du Cancer PRT-K 18-071, and the Fédération Leucémie espoir and Horizon Hemato.
Funding
The authors declare no competing financial interests or funding sources.
Author information
Authors and Affiliations
Contributions
C.B.: Data collection, methodology, analysis, validation, visualization, graphic representation, statistics, writing, original draft preparation; M.S.: Data collection, methodology, analysis, validation, visualization, graphic representation, statistics, reviewing and editing; N.G.: Reviewing and editing; L.L.: Analysis, reviewing and editing; A.T.: Reviewing and editing; G.A.: Reviewing and editing; J.B.: Reviewing and editing; E.L.: Reviewing and editing; A.P.: Reviewing and editing; N.B.: Resources, reviewing and editing; A.B.: Resources, reviewing and editing; Y.B.: Resources, reviewing and editing T.J.M.: Resources, reviewing and editing; V.A.: Conceptualization, methodology, validation, resources, reviewing and editing, supervision, project administration.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval/consent to participate
Diagnostic peripheral blood, bone marrow, or lymphoid tissue samples from 818 adults and children with T-LBLL (476 patients with T-ALL and 342 with T-LBL) were centrally analyzed in Necker-Enfants Malades Hospital (AP-HP, Paris France) after informed consent was obtained at diagnosis according to the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Bontoux, C., Simonin, M., Garnier, N. et al. Oncogenetic landscape of T-cell lymphoblastic lymphomas compared to T-cell acute lymphoblastic leukemia. Mod Pathol 35, 1227–1235 (2022). https://doi.org/10.1038/s41379-022-01085-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-022-01085-9
