
Abstract
Understanding the mechanisms of acquired resistance to anti-PD-1 will allow development of better treatment strategies for cancer patients. This study evaluated potential mechanisms of acquired resistance to anti-PD-1 in longitudinally collected metastatic melanoma patient biopsies. Thirty-four metastatic melanoma biopsies were collected from 16 patients who had initially responded to either anti-PD-1 (n=13) alone or combination of anti-PD-1 and ipilimumab (n=3) and then progressed. Biopsies were taken prior to treatment (PRE, n=12) and following progression of disease (PROG, n=22). Immunohistochemistry was performed on all biopsies to detect CD8, FOXP3, PD-1 and VISTA expression on T-cells and PTEN, β-catenin, PD-L1, HLA-A, and HLA-DPB1 expression in the tumor. The majority of patients showed significantly increased density of VISTA+ lymphocytes from PRE to PROG (12/18) (P=0.009) and increased expression of tumor PD-L1 from PRE to PROG (11/18). Intratumoral expression of FOXP3+ lymphocytes significantly increased (P=0.018) from PRE to PROG (10/18). Loss of tumor PTEN and downregulation of tumor HLA-A from PRE to PROG were each identified in 5/18 and 4/18 PROG biopsies, respectively. Downregulation of HLA-DPB1 from PRE to PROG was present in 3/18 PROG biopsies, whereas nuclear β-catenin activation was only identified in 2/18 PROG biopsies. Negative immune checkpoint regulation by VISTA represents an important potential mechanism of acquired resistance in melanoma patients treated with anti-PD-1. Downregulation of HLA-associated antigen presentation also occurs with acquired resistance. Augmentation of the VISTA immune checkpoint pathway may hold promise as a therapeutic strategy in metastatic melanoma patients, particularly those failing anti-PD-1 therapy, and warrants assessment in clinical trials.
Similar content being viewed by others


Main
The systemic treatment options for patients with metastatic melanoma have expanded significantly in the past decade. Inhibitors targeting the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4)1 and programmed cell death 1 (PD-1)2 receptors, either alone or in combination, have clinical efficacy and are now standard care, but long-term survival occurs only in a minority of patients due to innate and acquired resistance. In particular, acquired resistance, which is uncommon with CTLA-4 inhibitor ipilimumab monotherapy, is a frequent event with anti-PD-1 inhibitors, with 43% of patients initially responding to therapy subsequently progressing within 3 years.3 Understanding the biological mechanisms responsible for the development of acquired resistance will likely underpin effective new treatment strategies for such patients and is required to improve outcomes further.
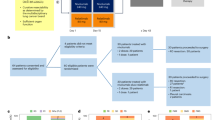
Predictive biomarkers of response to anti-PD-1 inhibitors, such as the expression of CD8+ and PD-1+ lymphocytes within the tumor and at the tumor–stromal interface, increased mutation burden, and PD-L1 expression by the tumor have recently been described by our group and others;4, 5, 6, 7 however, there are limited data describing the mechanisms that lead to acquired resistance with anti-PD-1 therapy. There are many proposed mechanisms based upon data derived from murine/cell line models of resistance but little information from human patients. Proposed mechanisms (Figure 1) include increased expression of non-redundant T-cell-inhibitory receptor pathways via VISTA (PD-1H),8, 9 LAG3, TIM3, and BTLA-4.10 Other potential mechanisms include upregulation of immunosuppressive ligands, such as PD-L1, by antigen-presenting cells (APCs) and tumor cells, and loss of MHC class I and II antigens preventing T-cell recognition.11, 12 JAK1 and JAK2 mutations resulting in dysfunctional interferon signaling have been shown to contribute to acquired resistance in a small subset of patients, and mutations to the antigen-processing gene B2M have also been implicated.13 Additional oncogene-driven immunosuppression may occur through loss of PTEN expression leading to activation of the PI3K/AKT pathway14 and increased WNT signaling through nuclear β-catenin.15

Schematic diagram representing the proposed mechanisms of resistance to immune checkpoint inhibitors evaluated in this study. Intracellular tumor-specific mechanisms of resistance include (1) loss/inactivation of the tumor-suppressor PTEN, (2) nuclear β-catenin activation, (3) PD-L1 loss or upregulation, and (4) loss of antigen presentation through downregulation of MHC-I and II. Immune microenvironment mechanisms include (5) lack of PD-1+ T-cells, (6) lack of CD8+ T-cells, (7) increased FOXP3+ regulatory T cells, and (8) expression of other co-inhibitory receptors, such as VISTA.
The aim of this study was to examine the expression of several inhibitory immune checkpoint receptors and HLA proteins in longitudinal samples from patients developing acquired resistance to immune checkpoint inhibitor therapy following an initial response. As far as we are aware, this is the first study to assess the combination of these potential mechanisms of acquired resistance in biopsies of metastatic melanoma in patients treated with immune checkpoint inhibitors.
Materials and methods
Study Design
Thirty-four biopsies from 16 patients were included in this study; the patients’ clinical characteristics are summarized in Table 1. All patients were treated with either anti-PD-1 inhibitor alone (nivolumab or pembrolizumab) or the combination of anti-PD-1 and anti-CTLA4 inhibitors (nivolumab or pembrolizumab with ipilimumab). All patients had a complete (n=4), partial response (n=10), or stable disease for 6 months duration (n=2) and then progressed with either a new lesion or an existing lesion that initially responded and progressed. Twenty-two tissue biopsies taken at disease progression (PROG) from 16 patients were available for analysis. Eighteen PROG biopsies from 12 patients also had a matched pretreatment (PRE) biopsy. All patients gave informed consent and all biopsies were conducted according to the Treat, Excise and Analyze for Melanoma protocol at the Melanoma Institute Australia (X11-0289, HREC/11/RPAH/444).16, 17
Immunohistochemistry
All immunohistochemical (IHC) staining was carried out on 4-μm-thick sections using an Autostainer Plus (Dako–Agilent Technologies) with appropriate positive and negative controls. Sections were baked for 60 min at 60 °C in a dehydration oven and heat-induced epitope retrieved in the PT link (Dako–Agilent Technologies) using either EnVision FLEX low pH (pH=6) or high pH (pH=9) target retrieval solution for 20 min at 97 °C and then cooled to room temperature in TBST wash buffer for 5 min. Slides were incubated with the following antibodies at the following dilutions: CD8 (Cell Marque, SP16) 1:200, PD-1 (Cell Marque, MRQ-22/NAT105) 1:100, FOXP3 (Abcam, AB22510) 1:200, VISTA (Cell Signaling, D1L2G) 1:200, PD-L1 (Cell Signaling, E1L3N) 1:200, β-catenin (Invitrogen, CAT5H10) 1:200, PTEN (Cell Signaling, 138G6) 1:200, HLA-A (Abcam, EP1395Y/ab52922) 1:400, and HLA-DPB1 (Abcam, Ab55152) 1:100. Antibody detection used the Envision FLEX Kit (K8023) with a DAB chromagen for visualization according to the manufacturer’s instructions (Dako–Agilent Technologies). Slides were then counterstained with hematoxylin.
Assessment of the immune markers (CD8, PD-1, FOXP3, VISTA) was undertaken using an immunoreactive score18 ranging from 0 to 300 as previously described,19, 20 made up of a four-tiered density score (0–3) multiplied by the percentage (0–100) of the tumor that the immune cells had infiltrated both at the interface of the tumor and the stroma (peritumoral) and within the tumor (intratumoral). Assessment of tumor markers (HLA-A, HLA-DPB1, PD-L1) was undertaken using an immunoreactive score18 ranging from 0 to 300 as previously described,19, 20 calculated using a four-tiered intensity score (0–3) multiplied by the percentage of tumor (0–100) expressing these markers. The expression of the tumor markers β-catenin and PTEN was assessed and scored as either present or absent in the nucleus for β-catenin and in the cytoplasm for PTEN. All IHC slides were independently reviewed (by LAJ and HK) and a consensus was reached on discrepant cases.
Statistical Analysis
Statistical analyses were conducted with ‘PASW Statistics 21’ SPSS, IBM. Wilcoxon matched-pairs method was used to test for significant changes in immune markers between the different biopsy time points (PRE and PROG). Correlations between the various immune markers were conducted using Spearman’s rho test.
Results
Patients and Melanoma Biopsies
Twenty-two PROG and 12 matched PRE biopsies from 16 patients treated with either anti-PD-1 inhibitor (n=13) or a combination of anti-PD-1 and anti-CTLA4 inhibitors (n=3) were examined. The clinicopathological and response characteristics of the patients are summarized in Table 1. Of the 22 PROG biopsies, 3 (14%) were existing lesions that originally shrank on treatment and then grew, and 19 (86%) were new lesions, ie, all PROG biopsies represented acquired resistance specimens. The median time to PROG biopsy from the commencement of therapy was 10.7 months (range 1.4–24.8 months). There were no significant correlations between clinicopathological features of the patients (ie, age, sex, mutation status, AJCC M-stage, LDH, ECOG performance status, time to PROG biopsy) with expression of any of the IHC markers at baseline or upon development of acquired resistance.
Resistance Mechanisms to Immune Checkpoint Inhibitors
Expression of intratumoral VISTA+ lymphocytes increased in 67% of PRE–PROG pairs (12/18), depicted in Figures 2 and 3a (P=0.009) and Figures 4a and b. The next most frequent finding was increased FOXP3+ Tregs (Figures 4c and d), seen in 56% (10/18) of PROG biopsies (P=0.018). Although the majority of PROG biopsies 61% (11/18) displayed an increase in tumoral PD-L1 (Figures 4g and h), the magnitude of the changes was generally small and the increase did not reach statistical significance (P>0.05).

Changes in immune checkpoint, expression, HLA expression and oncogenic signaling in relapsing metastatic melanoma patients following treatment with immune checkpoint inhibitors after an initial response. Each column represents an individual PROG biopsy and some patients had multiple PROG specimens. Patient columns are ordered based on average VISTA expression from right to left. Clinical data are depicted in the upper portion, change in the expression of markers from PRE to PROG in the middle and PROG expression of oncogenic markers in the bottom panel. Patients 5, 10, 13 and 14 did not have a matched PRE biopsy for analysis and are omitted from the figure. IT, intratumoral; nuc, nuclear expression; P, patient; PB, PROG biopsy; Q61K, NRASQ61K mutation present; Q61R, NRASQ61R mutation present; V600E, BRAFV600E mutation present; V600K, BRAFV600K mutation present; wild type, wild type for BRAF and NRAS.

The change in expression levels from PRE (prior to commencing treatment) to PROG (upon progression) in melanoma patients treated with immune checkpoint inhibitors. (a) There was a significant increase in the intratumoral expression of VISTA+ lymphocytes from PRE to PROG (P=0.009). (b) There was a significant increase in the intratumoral expression of FOXP3+ lymphocytes from PRE to PROG (P=0.018). *Significance taken at P<0.05.

Immunohistochemical assessment for immune cell-based mechanisms of resistance in metastatic melanoma patients treated with immune checkpoint inhibitors. (a and b) VISTA expression in paired biopsies from patient P8. (c and d) FOXP3 expression in paired biopsies from patient P1. (e and f) CD8+ T-cells in paired biopsies from patient P6. (g and h) PDL1 expression in paired biopsies from patient P1.
Loss of PTEN expression and decrease in tumor HLA-A and HLA-DPB1 expression were observed in 28% (5/18; Figures 5a and b), 22% (4/18; Figures 5e and f) and 17% (3/18; Figures 5g and h) of PROG biopsies compared with their matching PRE biopsies, respectively, while activation of nuclear β-catenin (Figures 5c and d) was detected in only 11% (2/18) of PROG biopsies. Although the non-PD-L1 oncogenic immunosuppression (as evident by downregulation of HLA and PTEN and upregulation of β-catenin expression) was less frequent, they tended to occur mutually exclusively in patients whose tumors demonstrated an increase in VISTA-expressing lymphocytes (Figure 3 and Table 2).

Immunohistochemical assessment for oncogenic mechanisms of resistance in metastatic melanoma patients treated with immune checkpoint inhibitors. (a and b) PTEN expression in paired biopsies from patient P6. (c and d) β-Catenin expression in paired biopsies from patient P16. (e and f) HLA-A expression in paired biopsies from patient P16. (g and h) HLA-DBP1 expression in paired biopsies from patient P7.
Melanoma and Immune Marker Correlates
Membranous tumor expression of PD-L1 was present (≥1% positivity) in 28 biopsies (82%) and significantly correlated with intratumoral CD8, FOXP3, PD-1, and VISTA expression (r=0.772, P=8.9 × 10−8; r=0.486, P=0.004; r=0.376, P=0.026; r=0.562, P=0.001, respectively; Table 2). The membranous tumor expression of HLA-A was significantly correlated with the tumor expression of HLA-DPB1 (r=0.42, P=0.015), and both markers inversely correlated with peritumoral VISTA+ lymphocytes (r=−0.434, P=0.01; r=−0.519, P=0.002, respectively; Table 2). Loss of nuclear β-catenin showed a weak association with increased levels of intratumoral CD8+ cell density (P=0.04). Membranous immune cell expression of VISTA was identified in all 34 specimens both within the tumor (intratumoral) and at the interface between the tumor and stroma (the peritumoral region, Figure 5). Intratumoral nuclear expression of FOXP3+ regulatory lymphocytes was strongly correlated with the VISTA expression (r=−0.620, P<0.001) but not with the PD-1 expression (P=0.251).
Discussion
This study is the first to describe the expression of VISTA-expressing lymphocytes in melanoma samples and in the context of acquired resistance to immune checkpoint inhibitors. The study highlights the need to better understand the receptor’s role in conferring resistance to immune checkpoint blockade and raises the possibility that VISTA inhibition may be an effective treatment strategy in melanoma patients.
The frequent observation of increased expression of intratumoral VISTA+ lymphocytes during acquired resistance to immune checkpoint inhibitor therapy suggests that this is probably an important mechanism of resistance. Therapeutic blockade of the VISTA pathway, possibly combined with anti-PD-1 inhibitors, represents a potentially efficacious treatment strategy in some patients, particularly as VISTA has been shown to non-redundantly regulate the activity of activated lymphocytes in murine models.21 The binding partner to VISTA has not yet been identified; however, it has been shown that it acts as both a ligand on APCs and as a receptor on activated infiltrating lymphocytes.8, 9 VISTA is known to suppress T-cell function as well as promote the differentiation of naive T-cells into Tregs22; this is supported in the current study by the strong correlation between changes in VISTA expression and Treg (FOXP3) density. Additionally, preclinical murine studies have shown that the combination of PD-L1 and VISTA blockade had a synergistic therapeutic effect in colon cancer models, with a lack of overt autoimmunity in VISTA/PD-1 double knockout mice which may offer a less toxic alternative to PD-1/CTLA-4 combination therapy.21
The downregulation of HLA molecules by the tumor is a mechanism used to evade recognition and killing by activated lymphocytes.23 Our study found a decrease in tumor HLA-A and HLA-DPB1 expression in 22% (4/18) and 17% (3/18) of PROG biopsies, which may negate the efficacy of tumor-infiltrating lymphocytes. Natural killer T-cells (NK-cells) are triggered by the loss of HLA expression that normally identifies infected, damaged, or transformed cells.24 Therefore, treatments that stimulate NK-cell function may overcome or prevent resistance in patients whose tumors evaded the immune system via HLA downregulation.25 Additionally, the use of interferon alone or in combination with a MEK1/2 inhibitor has been used to restore HLA expression and thereby increase antigenicity in a papillary thyroid carcinoma model12 and could potentially provide another avenue for combination therapy.
A loss of PTEN was seen in 28% (5/18) of PROG specimens. This is known to mediate resistance to immune checkpoint inhibitors through activation of the PI3K signaling pathway.14 Similarly, nuclear β-catenin, present in two patients’ PROG biopsies, has been shown to promote immune exclusion (T-cell and dendritic cells) through the WNT signaling pathway.26 Therefore, a proportion of patients may benefit from the combination of checkpoint inhibition with either PI3Kβ or FLT3 inhibitors (shown to counteract WNT immunosuppression), as they have been shown to increase efficacy with both anti-PD-1 and anti-CTLA-4 inhibitors in murine models.14
We observed an increase in tumor PD-L1 expression in the majority (11/18) of PROG specimens in relation to the matched PRE samples; however, this change was not significant. This probably reflects the fact that the magnitude of the change was often low (1%), which is partly a function of the dynamic nature of PD-L1 expression and highlights the pitfalls of using it as a biomarker.27 Nevertheless, the increase in PD-L1 expression was expected, as PD-1 blockade alone or in combination with CTLA-4 blockade promotes an increase in tumor-infiltrating lymphocytes28 and these activated lymphocytes produce interferon gamma that induces the expression of PD-L1 in the tumor cells,29 a finding confirmed in the current study with the strong positive correlation with CD8+ lymphocyte infiltration.
Acquired resistance to immune checkpoint inhibitors occurs through a number of mechanisms modulated by intracellular pathways and the tumor microenvironment. As was observed in three patients in the current study, multiple mechanisms probably contribute to the development of acquired resistance. Heterogeneity in mechanisms of resistance to immune checkpoint inhibitors in individual patients, as observed in the current study, highlights the difficulties associated with selecting the most appropriate subsequent treatment options for patients who develop acquired resistance to immune checkpoint inhibitor therapy.
References
Brunet J-F, Denizot F, Luciani M-F et al. A new member of the immunoglobulin superfamily—CTLA-4. Nature 1987;328:267–270.
Ishida Y, Agata Y, Shibahara K et al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 1992;11:3887.
Robert C, Ribas A, Hamid O et al. Three-year overall survival for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. J Clin Oncol 2016;34.
Tumeh PC, Harview CL, Yearley JH et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–571.
Vilain RE, Kakavand H, Menzies AM et al. PD1 inhibition-induced changes in melanoma and its associated immune infiltrate. Eur J Cancer 2015;51:S666.
Johnson DB, Frampton GM, Rioth MJ et al. Targeted next generation sequencing identifies markers of response to PD-1 blockade. Cancer Immunol Res 2016;4:959–967.
Apetoh L, Smyth MJ, Drake CG et al. Consensus nomenclature for CD8+ T cell phenotypes in cancer. Oncoimmunology 2015;4:e998538.
Wang L, Rubinstein R, Lines JL et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exp Med 2011;208:577–592.
Flies DB, Wang S, Xu H et al. A monoclonal antibody specific for the programmed death-1 homolog prevents graft versus host disease in mouse models. J Immunol 2011;187:1537–1541.
Restifo NP, Smyth MJ, Snyder A . Acquired resistance to immunotherapy and future challenges. Nat Rev Cancer 2016;16:121–126.
Johnson DB, Estrada MV, Salgado R et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun 2016;7:10582.
Angell TE, Lechner MG, Jang JK et al. MHC class I loss is a frequent mechanism of immune escape in papillary thyroid cancer that is reversed by interferon and selumetinib treatment in vitro. Clin Cancer Res 2014;20:6034–6044.
Zaretsky JM, Garcia-Diaz A, Shin DS et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Eng J Med 2016;375:819–829.
Peng W, Chen JQ, Liu C et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 2016;6:202–216.
Spranger S, Bao R, Gajewski TF . Melanoma-intrinsic [bgr]-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–235.
Long GV, Wilmott JS, Haydu LE et al. Effects of BRAF inhibitors on human melanoma tissue before treatment, early during treatment, and on progression. Pigment Cell Melanoma Res 2013;26:499–508.
Wilmott JS, Long GV, Howle JR et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res 2012;18:1386–1394.
Lade-Keller J, Rømer KM, Guldberg P et al. Evaluation of BRAF mutation testing methodologies in formalin-fixed, paraffin-embedded cutaneous melanomas. J Mol Diagn 2013;15:70–80.
Kakavand H, Wilmott JS, Menzies AM et al. PD-L1 expression and tumor-infiltrating lymphocytes define different subsets of MAPK inhibitor–treated melanoma patients. Clin Cancer Res 2015;21:3140–3148.
Kakavand H, Vilain RE, Wilmott JS et al. Tumor PD-L1 expression, immune cell correlates and PD-1+ lymphocytes in sentinel lymph node melanoma metastases. Mod Pathol 2015;28:1535–1544.
Liu J, Yuan Y, Chen W et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci USA 2015;112:6682–6687.
Le Mercier I, Chen W, Lines JL et al. VISTA regulates the development of protective antitumor immunity. Cancer Res 2014;74:1933–1944.
Lanier LL . NK cell recognition. Annu Rev Immunol 2005;23:225–274.
Boudreau Jeanette E, Liu X-R, Zhao Z et al. Cell-extrinsic MHC class I molecule engagement augments human NK cell education programmed by cell-intrinsic MHC class I. Immunity 2016;45:280–291.
Romagne F, Andre P, Spee P et al. Preclinical characterization of 1-7F9, a novel human anti-KIR receptor therapeutic antibody that augments natural killer-mediated killing of tumor cells. Blood 2009;114:2667–2677.
Spranger S, Gajewski TF . A new paradigm for tumor immune escape: beta-catenin-driven immune exclusion. J Immunother Cancer 2015;3:43.
Madore J, Strbenac D, Vilain R et al. PD-L1 negative status is associated with lower mutation burden, differential expression of immune-related genes, and worse survival in stage III melanoma. Clin Cancer Res 2016;22:3915–3923.
Curran MA, Montalvo W, Yagita H et al. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA 2010;107:4275–4280.
Krönig H, Kremmler L, Haller B et al. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur J Haematol 2013;92:195–203.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
GL is a consultant advisor to Amgen, Array, BMS, Merck Sharpe & Dohme, Oncosec, Pierre Fabre, Novartis and Roche and has received Honoraria from BMS, Merck Sharpe & Dohme, Novartis and Roche. AMM is a consultant advisor to MSD, Novartis, Pierre Fabre and Chugai and has received honoraria from BMS and Roche. MSC is a consultant advisor for MSD, BMS and Novartis and has received honoraria from MSD and Novartis. RPMS has received Honoraria from BMS. The other authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Kakavand, H., Jackett, L., Menzies, A. et al. Negative immune checkpoint regulation by VISTA: a mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod Pathol 30, 1666–1676 (2017). https://doi.org/10.1038/modpathol.2017.89
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.89
This article is cited by
-
The role of circular RNAs in regulating resistance to cancer immunotherapy: mechanisms and implications
Cell Death & Disease (2024)
-
Overcoming acquired resistance to cancer immune checkpoint therapy: potential strategies based on molecular mechanisms
Cell & Bioscience (2023)
-
VISTA and its ligands: the next generation of promising therapeutic targets in immunotherapy
Cancer Cell International (2023)
-
The molecular and functional landscape of resistance to immune checkpoint blockade in melanoma
Nature Communications (2023)
-
Recent advancements in the B7/CD28 immune checkpoint families: new biology and clinical therapeutic strategies
Cellular & Molecular Immunology (2023)
