
Abstract
Chimeric antigen receptor (CAR)-T cell therapy is a revolutionary new pillar in cancer treatment. Although treatment with CAR-T cells has produced remarkable clinical responses with certain subsets of B cell leukemia or lymphoma, many challenges limit the therapeutic efficacy of CAR-T cells in solid tumors and hematological malignancies. Barriers to effective CAR-T cell therapy include severe life-threatening toxicities, modest anti-tumor activity, antigen escape, restricted trafficking, and limited tumor infiltration. In addition, the host and tumor microenvironment interactions with CAR-T cells critically alter CAR-T cell function. Furthermore, a complex workforce is required to develop and implement these treatments. In order to overcome these significant challenges, innovative strategies and approaches to engineer more powerful CAR-T cells with improved anti-tumor activity and decreased toxicity are necessary. In this review, we discuss recent innovations in CAR-T cell engineering to improve clinical efficacy in both hematological malignancy and solid tumors and strategies to overcome limitations of CAR-T cell therapy in both hematological malignancy and solid tumors.
Similar content being viewed by others
Introduction
Chimeric antigen receptor (CAR)-T cell therapy has been revolutionary as it has produced remarkably effective and durable clinical responses1. CARs are engineered synthetic receptors that function to redirect lymphocytes, most commonly T cells, to recognize and eliminate cells expressing a specific target antigen. CAR binding to target antigens expressed on the cell surface is independent from the MHC receptor resulting in vigorous T cell activation and powerful anti-tumor responses2. The unprecedented success of anti-CD19 CAR-T cell therapy against B cell malignancies resulted in its approval by the US Food and Drug Administration (FDA) in 20173,4,5. However, there are major limitations to CAR-T cell therapy that still must be addressed including life-threatening CAR-T cell-associated toxicities, limited efficacy against solid tumors, inhibition and resistance in B cell malignancies, antigen escape, limited persistence, poor trafficking and tumor infiltration, and the immunosuppressive microenvironment. In addition, the workforce must adapt to meet the needs of this growing and evolving field by developing educational programs to train a workforce6. Many approaches including combining CAR-T cell therapy with other anticancer therapies or employing innovative CAR engineering strategies to improve anti-tumor efficacy, expand clinical efficacy, and limit toxicities have been proposed. In this review, we discuss recent innovations in CAR-T cell engineering to improve clinical efficacy in both hematological malignancy and solid tumors and strategies to overcome current limitations (Table 1), including antigen escape, CAR-T cell trafficking, tumor infiltration, the immunosuppressive microenvironment, and CAR-T cell-associated toxicities (Fig. 1).

Current challenges in CAR-T cell therapy include (A) antigen escape, (B) on-target off-tumor effects, (C) trafficking and infiltration of tumors, (D) the immunosuppressive tumor microenvironment, and (E) CAR-T cell-associated toxicities.
CAR Structure
CARs are modular synthetic receptors that consist of four main components: (1) an extracellular target antigen-binding domain, (2) a hinge region, (3) a transmembrane domain, and (4) one or more intracellular signaling domains. Here we will discuss the current principles underlying CAR design.
Antigen binding domain
The antigen binding domain is the portion of the CAR that confers target antigen specificity. Historically, the antigen-binding domains are derived from the variable heavy (VH) and light (VL) chains of monoclonal antibodies, connected via a flexible linker to form a single-chain variable fragment (scFv). Classically, the scFvs present in CARs target extracellular surface cancer antigens resulting in major histocompatibility complex (MHC)-independent T cell activation, although recognition of intracellular tumor-associated antigens using MHC-dependent, T cell receptor (TCR)-mimic CARs have been described7. Several characteristics of the scFv impact CAR function beyond simply recognizing and binding the target epitope. For instance, the mode of interaction among the VH and VL chains as well as the complementarity-determining regions’ relative positions impact the affinity and specificity of the CAR for its target epitope8. Affinity is a particularly important antigen-binding domain parameter as it fundamentally determines CAR function. In order to recognize antigens on tumor cells, induce CAR signaling, and activate T cells, the CARs antigen binding affinity must be sufficiently high but not high enough to result in activation induced death of the CAR expressing T cell and trigger toxicities (discussed later in this review)9,10. While affinity is certainly one of the most important factors to further complicate matters, it has been shown that even scFvs with similar affinities can differentially impact CAR-T cell function. Therefore, in order to optimize binding of the CAR to its target antigen, additional factors such as epitope location, target antigen density, and avoidance of scFvs associated with ligand-independent tonic signaling must be considered.
Hinge region
The hinge or spacer region is defined as the extracellular structural region that extends the binding units from the transmembrane domain. The hinge functions to provide flexibility to overcome steric hindrance and contributes to the length in order to allow the antigen-binding domain to access the targeted epitope. Importantly, the selected hinge appears to impact CAR functionality as differences in the length and composition of the hinge region can affect flexibility, CAR expression, signaling, epitope recognition, strength of activation outputs, and epitope recognition11,12. In addition to these affects, it has been proposed that the spacer length is critical to provide sufficient intercellular distance to allow for immunological synapse formation13. In principle, the “optimal” spacer length is dependent on the position of the target epitope and the level of steric hindrance on the target cell in which long spacers provide added flexibility and allow more effective access to membrane-proximal epitopes or complex glycosylated antigens, while short hinges are more successful at binding membrane-distal epitopes11,14,15,16. In practice, however, the proper spacer length is often determined empirically and must be tailored for each specific antigen-binding domain pair. There are numerous examples in the literature of short spacer CARs (CD19 and carcinoembryonic antigen (CEA))14 and long spacer CARs (mucin 1 (MUC1)), membrane-proximal epitopes of receptor tyrosine kinase-like orphan receptor 1 (ROR1)16. The most commonly employed hinge regions are derived from amino acid sequences from CD8, CD28, IgG1, or IgG4. IgG-derived spacers, however, can cause CAR-T cell depletion and thus, decreased persistence in vivo as they can interact with Fcγ receptors17,18. These effects can be avoided by either the selection of a different spacer region or through additional engineering of the spacer region based on functional or structural considerations.
Transmembrane domain
Among all of the components of CAR’s, the transmembrane domain is probably the least characterized region. The major function of the transmembrane domain is to anchor the CAR to the T cell membrane, although evidence suggests that the transmembrane domain can also be relevant for CAR-T cell function19,20. More specifically, studies suggest that the CAR transmembrane domains influence CAR expression level, stability, can be active in signaling or synapse formation, and dimerize with endogenous signaling molecules19,20,21. Most transmembrane domains are derived from natural proteins including CD3ζ, CD4, CD8α, or CD28. The effect of one transmembrane compared to another on CAR function is not well studied as the transmembrane domain is frequently changed based on the requirements of the extracellular spacer region or the intracellular signaling domains. Notably, the CD3ζ transmembrane may facilitate CAR-mediated T cell activation as the CD3ζ transmembrane domain mediates CAR dimerization and incorporation into endogenous TCRs19. These beneficial effects of the CD3ζ transmembrane domain come at the cost of decreasing CAR stability compared to CAR’s with the CD28 transmembrane domain22. Together, the impact of the transmembrane domain and the hinge region appear to also influence CAR-T cell cytokine production and activation induced cell death (AICD) as CAR-T cells with CD8α transmembrane and hinge domains release decreased amounts of TNF and IFNγ and have decreased susceptibility to AICD compared to CARs with these domains derived from CD2823. Overall, studies suggest that proper CAR-T cell signaling may be best facilitated by linking the proximal intracellular domain to the corresponding transmembrane domain, while CAR expression and stability may be enhanced by using the frequently used CD8α or CD28 transmembrane domains.
Intracellular signaling domain(s)
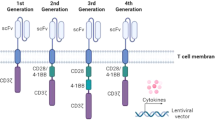
Arguably the most attention in CAR engineering has been focused on understanding the effects of CAR co-stimulation with the goal of generating CAR constructs with the optimal endodomain. First generation CARs engineered in the late 1990s contained a CD3ζ or FcRγ signaling domain24. A large majority of CARs rely on activation of CAR-T cells through CD3ζ derived immunoreceptor tyrosine-based activation motifs25. Effective T cell responses are not able to be generated by only signaling with these motifs however26. The durability and persistence of these first generation CARs are not robust in vitro26. These findings were echoed by clinical studies that showed limited or no efficacy27,28.
The importance of co-stimulation in CD-19-targeted CAR-T cell persistence was demonstrated using early in vivo models of B-cell malignancies29. IL-2 production and proliferation upon repeated antigen exposure were improved by adding a co-stimulatory domain30. With this understanding of the importance of co-stimulation for durable CAR-T cell therapy, second generation CARs with one co-stimulatory domain in series with the CD3ζ intracellular signaling domain were generated30,31. The two most common, FDA-approved co-stimulatory domains CD28 and 4-1BB (CD137) are both associated with high patient response rates. The co-stimulatory domains differ in their functional and metabolic profiles in which CARs with CD28 domains differentiate into effector memory T cells and primarily use aerobic glycolysis while CARs possessing the 4-1BB domain differentiate into central memory T cells and display increased mitochondrial biogenesis and oxidative metabolism32. Clinically, second generation CAR-T cells have produced strong therapeutic responses in several hematological malignancies, including chronic lymphocytic leukemia, B-cell acute lymphoblastic leukemia, diffuse large B-cell lymphoma, and multiple myeloma and the efficacy of second generation CAR-T cells are currently being investigated in solid tumors, including glioblastoma, advanced sarcoma, liver metastases, as well as mesothelioma, ovarian cancer, and pancreatic cancer33. Several alternative co-stimulatory domains such as inducible T cell co-stimulator (ICOS)34, CD27 (ref. 35), MYD88 and CD40 (ref. 36), and OX40 (CD134) (ref. 37) have demonstrated preclinical efficacy although clinical investigation is still pending. It has been hypothesized that co-stimulation through only one domain produces incomplete activation, resulting in the production of third generation CARs, which incorporate two costimulatory domains in series with CD3ζ38. Preclinical studies of third generation CARs have produced mixed results. Specifically, CARs incorporating CD28 and 4-1BB signaling resulted in stronger cytokine production in lymphoma, and pulmonary metastasis showed an improved in vivo antitumor response compared to second generation CARs39. In leukemia and pancreatic cancer models, third generation CARs showed no in vivo treatment benefits and failed to outperform second generation CARs in their respective models40,41.
Limitations of CAR-T cell therapy
Antigen escape
One of the most challenging limitations of CAR-T cell therapy is the development of tumor resistance to single antigen targeting CAR constructs. Although initially single antigen targeting CAR-T cells can deliver high response rates, the malignant cells of a significant portion of patients treated with these CAR-T cells display either partial or complete loss of target antigen expression. This phenomenon is known as antigen escape. For example, although 70–90% of relapsed and/or refractory ALL patients show durable responses to CD19 targeted CAR-T cell therapy, recent follow-up data suggest development of a common disease resistance mechanism, including downregulation/loss of CD19 antigen in 30–70% of patients who have recurrent disease after treatment42,43. Similarly, downregulation or loss of BCMA expression in multiple myeloma patients being treated with BCM targeted CAR-T cells has been observed44,45,46. Similar antigen escape resistance patterns have been observed in solid tumors. For example, a CAR-T cell therapy case report that targeted IL13Ra2 in glioblastoma suggested that tumor recurrences displayed decreased IL13Ra2 expression47. In order to reduce the relapse rate in CAR-T cell treatment of both hematological malignancies and solid tumors, many strategies are now relying on targeting multiple antigens. These employ the use of either dual CAR constructs or tandem CARs, which is a single CAR construct that contains two scFvs in order to concomitantly target multiple target tumor antigens. Clinically, it appears that both of these strategies may result in prolonged durable remission rates, and there are several CD19 and CD20 or CD 19 and CD22 clinical trials25. Excitingly, preliminary results from clinical trials using dual-targeted CAR-T cells (CD19/CD22 or CD19/BCMA) have demonstrated promising results48,49,50,51. More specifically, preliminary clinical trial results of CD19/CD22 CAR-T cell therapy have demonstrated promising efficacy in adult patients with ALL and diffuse large B cell lymphoma50,51. Furthermore, preliminary results of BCMA/CD19 targeted CARs in the treatment of multiple myeloma suggest BCMA/CD19 targeted CARs are highly efficacious with favorable safety profiles48,49. In solid tumors, several tandem CARs have been tested in preclinical models including HER2 and IL13Ra2 in glioblastoma and HER2 and MUC1 in breast cancer. In both cases, dual targeting resulted in superior anti-tumor responses compared to single targeted therapy28,52. In the glioblastoma study, CARs targeting HER2 and IL13Ra2 led to improved anti-tumor activity and decreased antigen escape when compared against two other dual-targeting therapies53. This study illustrates the importance of optimizing the selection of target antigens that not only improve antitumor response but also decrease antigen escape mechanisms to prevent relapse.
On-target off-tumor effects
One of the challenges in targeting solid tumor antigens is that solid tumor antigens are often also expressed on normal tissues at varying levels. Therefore, antigen selection is crucial in CAR design to not only ensure therapeutic efficacy but also to limit “on-target off-tumor” toxicity. A potential avenue to overcome the targeting of antigens on solid tumors that are also present on normal tissues is the targeting of tumor-restricted post-translational modifications such as solid tumor overexpressed truncated O-glycans such as Tn (GalNAca1-O-Ser/Thr) and sialyl-Tn (STn) (NeuAca2–6-GalNAca1-O-Ser/Thr)54. Four major CAR-T cell targets have been investigated including TAG7228, B7-H3 (refs. 55,56), MUC1 (ref. 16), and MUC16 (refs. 57,58). Although first generation CAR-T cells targeting TAG72 in colorectal cancer produced no anti-tumor response, new versions of second generation TAG72-CAR-T cells and other tumor-restricted post-translational modifications are currently being investigated28,59. Further development of innovative strategies to reduce antigen escape and select antigens capable of inducing a sufficient antitumor efficacy, while minimizing toxicity concerns will be necessary in order to expand the clinical use of CAR-T cell therapies in hematological malignancy and solid tumors.
CAR-T cell trafficking and tumor infiltration
Compared to hematological malignancies, solid tumor CAR-T cell therapy is limited by the ability of CAR-T cells to traffic to and infiltrate solid tumors as the immunosuppressive tumor microenvironment and physical tumor barriers such as the tumor stroma limit the penetration and mobility of CAR-T cells. One strategy to ameliorate these limitations is through the utilization of delivery routes other than systemic delivery as local administration (1) eliminates the need for CAR-T cells to traffic to disease sites and (2) limits on-target off-tumor toxicities as the CAR-T cells’ on-target activity is directed on tumor cells minimizing interaction with normal tissues60. Preclinical models have demonstrated superior therapeutic efficacy of intraventricular injection of CAR-T cells targeting HER2 (ref. 61) and IL13Ra2 (ref. 62) in breast cancer brain metastases and in glioblastoma. These studies have led to three ongoing clinical trials investigating intraventricular injection of CAR-T cells in glioblastoma (NCT02208362, NCT03389230) and recurrent brain or leptomeningeal metastases (NCT03696030). Similarly, preclinical models showed superior CAR-T cell treatment of malignant pleural mesothelioma through intrapleural injection, which has resulted in an ongoing phase 1 clinical trial (NCT02414269) (ref. 63). Although localized injection appears to have superior efficacy, theoretically this approach is limited to single tumor lesions/oligometastatic disease25.
One recently developed strategy that appears to significantly improve CAR-T cell trafficking involves expressing chemokine receptors on CAR-T cells that match and respond to tumor-derived chemokines64. For example, recent studies have demonstrated that integrin αvβ6-CAR-T cells modified to express CXCR2 or CAR-T cells overexpressing CXCR1 or CXCR2 both enhance trafficking and significantly improve antitumor efficacy64,65,66. Physical barriers such as the tumor stroma also limit CAR-T cell therapy as these physical barriers prevent tumor penetration. Stroma is mostly composed of extracellular matrix in which heparin sulfate proteoglycan (HSPG) is the primary component that CAR-T cells must degrade in order to pass into the tumor67. CAR-T cells that have been engineered to express heparanase, an enzyme that degrades HSPG, show enhanced tumor infiltration and antitumor activity68. Similarly, fibroblast activation protein (FAP)-targeted CAR-T cells demonstrate increased cytotoxic function through reducing tumor fibroblasts in animal models69. In the future, there is a need for the development of innovative delivery strategies and approaches to improve tumor penetration in order to extend therapeutic efficacy to complex solid tumors and metastases.
Immunosuppressive microenvironment
In the tumor microenvironment, many cell types that drive immunosuppression can infiltrate solid tumors including myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs)70. These infiltrates and tumor cells drive the production of tumor facilitating cytokines, chemokines, and growth factors. In addition, immune checkpoint pathways such as PD-1 or CTLA-4 can serve to decrease antitumor immunity. One of the main causes of no response or a weak response to CAR-T cell therapy is poor T cell expansion and short-term T cell persistence. It has been hypothesized that development of this T cell exhaustion is triggered by co-inhibitory pathways71. Therefore, combination immunotherapy with CAR-T cells and checkpoint blockade is thought to be the next immunotherapy frontier as it provides the two elements necessary for strong immune responses:1 CAR-T cells, which provide the infiltrate and2 PD-1/PD-L1 blockade, which can ensure sustained T cell persistence and function72. In hematological malignancy, combination PD-1 blockade and CD19 CAR-T cell therapy in 14 children with heavily pretreated B-ALL resulted in improved persistence of CAR-T cells and better outcomes at a single-center study at Children’s Hospital of Pennsylvania73. In solid tumors, there are currently many studies aiming to evaluate the response rate of combination therapy71,74. One intriguing study in which 11 mesothelioma patients who received preconditioning with cyclophosphamide followed by a single dose of mesothelin targeted CAR-T cells and at least three doses of anti-PD-1agent resulted in a 72% response rate and complete metabolic responses in two patients75. Combining other forms of immunotherapy strategies may still be necessary in order to combat the inhibitory signal present in the tumor microenvironment.
Recently efforts have focused on engineering CARs that are resistant to immunosuppressive factors in the hostile tumor microenvironment such as TGF β-mediated inhibitory signals76. Another intriguing strategy involves the engineering of CAR-T cells to provide immunostimulatory signals in the form of stimulatory cytokines that increase survival, proliferation, antitumor activity of T cells, and rebalance the tumor microenvironment77. Many studies have investigated numerous cytokines to create these “armored CARs”. Studies that focus on the expression of pro-inflammatory cytokines instead of focusing on inhibitory signals have relied on IL-12 secretion78, IL-15 expression79, and redirecting immunosuppressive cytokines (e.g., IL-4) signaling towards proinflammatory cytokines80.
Although combination checkpoint blockade-CAR-T cell therapy is likely a new immunotherapy option, it is important to also recognize that even this combination may still be insufficient to induce infiltration of T cells and effector function. Therefore, additional studies combining CAR-T cell therapy and checkpoint blockade with other immunotherapies/strategies may be necessary to result in T cell infiltration and effector function in complex hematological malignancies or solid tumors.
CAR-T cell-associated toxicities
Although CAR-T cell therapy has been a revolutionary cancer treatment tool, high rates of toxicities with some fatalities have prevented CAR-T cell therapy from becoming first-line treatment. Critical factors that likely determine the incidence and severity of CRS, HLH/MAS, and/or ICANS are the design of the CAR, the specific target, and the tumor type81. To date, the toxicities underlying CAR-T cell therapy have been most extensively characterized in patients receiving the first FDA approved CAR-T cell therapy, CD19-directed CARs82,83. Even in the clinical trials with the most dramatic response rates, severe, life-threatening events have occurred in patients4,5,84. Specifically, in the case of acute lymphoblastic leukemia/lymphoma (ALL/LBL) patients treated with CAR-T cell therapy, nearly all patients have at least some less severe toxicity manifestations while 23–46% of patients displayed severe supraphysiologic cytokine production and massive in-vivo T cell expansion85. These toxic levels of systemic cytokine release and severe immune cell cross-activation in some patients result in the following toxicities:1 cytokine-release syndrome (CRS), which is associated with supraphysiologic cytokine production and massive in vivo T cell expansion2 hemophagocytic lymphohistiocytosis and/or macrophage activation syndrome (MAS) defined as a severe hyperinflammatory syndrome characterized by CRS and combinations of elevated serum ferritin and hemophagocytosis, renal failure, liver enzymes, splenomegaly, pulmonary edema, and/or absence of NK cell activity, and3 immune effector cell-associated neurotoxicity syndrome (ICANS), which is characterized by elevated cerebrospinal fluid cytokine levels and blood–brain barrier disruption86.
Mechanistically, CRS is a result of administered CAR-T cells becoming extensively activated resulting in the release of massive amounts of cytokines. Clinical manifestations of mild CRS is fever accompanied by fatigue, diarrhea, headache, rashes, arthralgia, and myalgia and in more severe cases, patients may present with hypotension, cardiac dysfunction, circulatory collapse, respiratory failure, renal failure, multiorgan system failure, and with possible progression to death3,4,87. In total, 77–93% of patients with leukemia receiving CAR-T cell therapy and 37–93% of patients with lymphoma receiving CAR-T cell therapy had any grade of CRS while 46% of patients treated with tisagenlecleucel for relapsed/refractory B-ALL and 13–18% of patients treated with axicabtagene ciloleucel and tisagenlecleucel, respectively for diffuse large B-cell lymphoma had ≥Grade 3 CRS3,4. Pathophysiologically, CRS is believed to be primarily mediated by IL-6 and therefore, management relies on the use of IL-6 receptor blockade with tocilizumab and corticosteroids3,4,5. Even with the use of tocilizumab, which is FDA approved to treat severe CRS, severe CRS and death still occur. Interestingly, HLH/MAS secondary to CAR-T cell therapy can be refractory to IL-6 inhibition and instead may require chemotherapy. While the incidence of HLH/MAS secondary to CAR-T cell therapy is unclear due to overlap with high-grade CRS, it has been reported in ≈1% of patients receiving CAR-T cell therapy88. In the case of neurotoxicity, the underlying pathophysiology and mechanisms are not completely understood88. Clinical manifestations of ICANS range from confusion, headache, attention deficits, word-finding difficulties, focal neurological deficits, or encephalopathy to life-threatening cerebral edema, transient coma, or seizures89. Neurotoxicity following CAR-T cell therapy is relatively common and can occur in up to 67% and 62% of patients receiving treatment for leukemia and lymphoma, respectively86. Management of neurotoxicity focuses on corticosteroids as IL-6 inhibitors are often not effective for neurotoxicity associated with CAR-T cell therapy90,91. To date, there remain no approved therapies for the prevention of the above toxicities, making it essential to optimize CAR engineering and employ other strategies to decrease CAR-induced toxicities88. Below, we review lessons learned in engineering CARs to reduce toxicity and additional strategies to ameliorate toxicities in CAR-T cell therapy.
Engineering CAR-T cells to ameliorate toxicity
In order to achieve efficacious therapeutic responses, a CAR-T cell antigen-binding domain must bind its target epitope and reach a minimum threshold level to induce CAR-T cell activation and cytokine secretion. At the same time, however, there is also some threshold level of activation that when surpassed produces toxic levels of cytokines and immune system activation. In other words, the CAR-T cell must remain within its therapeutic window to be clinically effective as overshooting the therapeutic window will lead to toxicity. From an engineering perspective, the degree of CAR-T cell activation and activation kinetics are influenced by several factors including but not limited to the level of tumor antigen expressed on malignant cells, tumor burden, antigen binding domain’s affinity to its target epitope, and the CAR’s costimulatory elements33,92. Therefore, careful consideration of several components of the CAR’s modular structure is necessary to optimize therapeutic efficacy and limit toxicity.
Altering CAR structure
One route to decrease toxicity is through altering the affinity of the CAR-T cell’s antigen binding domain. Decreasing the affinity of the antigen-binding domain would be expected to result in an increased requirement for higher antigen density on tumor cells in order for high levels of activation to be achieved. Therefore, it would be expected that decreased antigen affinity would circumvent the targeting of healthy tissue with a relatively low amount of antigen. Studies investigating this rationale have demonstrated that antigen-binding domains with micromolar affinity were much more selective for tumors with higher levels of target antigen expression compared to antigen binding domains with low nanomolar/sub-nanomolar affinity9.
It is also possible to modulate cytokine secretion via activated CAR-T cells by modifying the hinge and transmembrane regions. For instance, in a CD19-targeted CAR, modification of the CD8-α derived hinge and transmembrane amino acid sequences led to lower levels of cytokine release and decreased CAR-T cell proliferation93. Optimizing the hinge and transmembrane regions could be a useful approach to decrease toxicity as in phase 1 clinical trial, these modified hinge and transmembrane region CARs resulted in complete remission in 54.5% of B cell lymphoma patients (6/11 patients), and importantly, there were no CRS or ICANS events grade >193.
The costimulatory domain offers another modifiable region in CAR design that can be tailored based on tumor type, tumor burden, antigen density, target antigen–antigen binding domain pair, and concerns of toxicity. Specifically, 4-1BB domains result in a lower risk of toxicities, higher T cell endurance, and a lower peak level of T cell expansion, while CD28 co-stimulatory domains are associated with CAR-T cell activity that is more rapid in onset and subsequent exhaustion94. Therefore, 4-1BB costimulatory domains, which produce less toxicity may be particularly useful in cases where there is a high disease burden and/or a high antigen density tumor, while CD28 costimulatory domains may be necessary in order to achieve the required T cell activation threshold in cases where there is low total surface antigen density and/or a low-affinity antigen binding domain CAR94.
CAR Immunogenicity
The recognition of CAR constructs by the host immune system may contribute to cytokine-related toxicities and thus, utilizing human or humanized antibody fragments instead of murine-derived CARs to decrease CAR immunogenicity may be advantageous25,95. In addition, the hinge and/or transmembrane domains can be modified in order to decrease the immunogenicity of CAR, and also interestingly CAR-T cell persistence is improved95,96.
Modifying CAR transduced T cells and neurotoxicity
An exciting, recently developed avenue to prevent CAR-T cell cytokine toxicities is based on modifying the CAR transduced T cells. Cytokines and myeloid cells appear to play a significant role in CAR-T cell induced neurotoxicity as reports have shown significant increases of CD14+ cells in patients with grade 3 or higher neurotoxicity97 and a pivotal large B cell lymphoma CAR-T cell clinical trial showed that among serum biomarkers associated with development with grade 3 or higher neurotoxicity, GM-CSF elevation was most significantly associated with neurotoxicity3. Recent preclinical studies have demonstrated that neurotoxicity and CRS are decreased and CAR-T cell activity is increased after inhibition of macrophage activating and monocyte activating cytokine GM-CSF with lenzilumab87,98,99. GM-CSF mutational inactivation also appears to have similar effects in CAR transduced T cells98,100.
Therefore, these findings suggest that GM-CSF neutralization helps diminish neurotoxicity and reduce CRS98. In addition, deletion of tyrosine hydroxylase in a myeloid cell specific manner or inhibition of this enzyme using metyrosine results in decreased catecholamine and cytokine levels101. Preclinical evidence also suggests that IL-1 receptor antagonists reduced a form of neuroinflammation in leukemia/lymphoma mouse models treated with CD19 targeted CARs102.
CAR “off-switches”
Another potential avenue to ameliorate CAR-T cell toxicity is through implementing “off-switches” or suicide gene strategies. Such strategies would facilitate the ability to selectively decrease engineered cells at the onset of adverse events through the treatment with a secondary inducing agent103. Several approaches utilizing these concepts have been developed. For instance, independent expression or CAR constructs engineered to express full length CD20 or CD20 mimotopes facilitate the depletion of CAR-T cells via treatment with rituximab104. A limitation with this approach, however, is the relatively slow onset of antibody-mediated depletion of CAR-T cells may limit the efficacy of this approach in patients that require immediate reversal during severe, acute cytokine-mediated toxicities. This led to the impetus to develop faster switches such as inducible cas9, which in a clinical trial eliminated >90% of engineered T cells within 30 min105. Other strategies have relied on protease-based small molecule-assisted shutoff CARs (SMASh-CARs), which are also referred to as switch-off CARS (SWIFF-CARs)106. The biggest limitation with suicide strategies or other similar approaches is that although they are attractive for ensuring safety, their use abruptly stops therapy for rapidly progressing disease. This limitation has served as a strong incentive to develop strategies to ensure safety while leaving suicide gene activation as the last resort. One approach with exciting potential involves the use of dasatinib, a tyrosine kinase inhibitor, which functions to suppress the activation of T cells through inhibiting proximal TCR signaling kinases107. In preclinical models, dasatinib quickly and reversibly prevents the activation of CAR-T cells, and administration of dasatinib early after CAR-T cell infusion results in a significant mortality reduction of mice from otherwise fatal CRS107. Thus, this approach appears to provide temporary inhibition of CAR-T cell function and could allow for the rescue of CAR-T cell therapy after toxicities have subsided. In the future, the development of additional innovative approaches that temporarily inhibit CAR-T cell function and allow for CAR-T cell therapy rescue once the toxicity subsides will be necessary for CAR-T cell therapy to move towards first-line therapy for both hematological malignancy and solid tumors.
Conclusions
CARs are modular synthetic receptors that consist of four main components: an extracellular target antigen-binding domain, a hinge region, a transmembrane domain, and one or more intracellular signaling domains. CAR-T cells have revolutionized the treatment of certain hematological malignancies. However, obstacles still remain, which were discussed in this review. Training a workforce to meet the demands of this complex and evolving field is challenging and requires innovative curriculum development6. Antigen selection is critical to CAR-T cell function. Tumor cells can downregulate antigens due to the selective pressure of the CAR-T cells. Even with appropriate antigen targeting, on-target off-tumor effects can occur and cause associated toxicity. In solid tumors, getting CAR-T cells to traffic to and infiltrate the tumor is a challenge. This obstacle can be compounded by the immunosuppressive microenvironment of malignancies. Effective treatment also runs the risk of CAR-T cell-associated toxicities such as CRS and neurotoxicity. However, while there are challenges, new strategies and potential solutions continue to evolve and may provide a path forward to more effective and safer future therapies.
References
June, C. H., O’Connor, R. S., Kawalekar, O. U., Ghassemi, S. & Milone, M. C. C. A. R. T cell immunotherapy for human cancer. Science. 359, 1361–1365 (2018).
Sadelain, M., Brentjens, R. & Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 3, 388–398 (2013).
Neelapu, S. S. et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 377, 2531–2544 (2017).
Maude, S. L. et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 378, 439–448 (2018).
Schuster, S. J. et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N. Engl. J. Med. 377, 2545–2554 (2017).
Sterner, R. M. et al. A graduate-level interdisciplinary curriculum in CAR-T cell therapy. Mayo. Clin. Proc. Innov. Qual. Outcomes. 4, 203–210 (2020).
Zhang, G. et al. Anti-melanoma activity of T cells redirected with a TCR-like chimeric antigen receptor. Sci. Rep. 4, 1–8 (2014).
Chailyan, A., Marcatili, P. & Tramontano, A. The association of heavy and light chain variable domains in antibodies: implications for antigen specificity. FEBS J. 278, 2858–2866 (2011).
Liu, X. et al. Affinity-Tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 75, 3596–3607 (2015).
Caruso, H. G. et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 75, 3505–3518 (2015).
Hudecek, M. et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 3, 125–135 (2015).
Jensen, M. C. & Riddell, S. R. Designing chimeric antigen receptors to effectively and safely target tumors. Curr. Opin. Immunol. 33, 9–15 (2015).
Srivastava, S. & Riddell, S. R. Engineering CAR-T cells: design concepts. Trends Immunol. 36, 494–502 (2015).
Guest, R. D. et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J. Immunother. 28, 203–211 (2005).
James, S. E. et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J. Immunol. 180, 7028–7038 (2008).
Wilkie, S. et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J. Immunol. 180, 4901–4909 (2008).
Hombach, A., Hombach, A. A. & Abken, H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc “spacer” domain in the extracellular moiety of chimeric antigen receptors avoids “off-target” activation and unintended initiation of an innate immune response. Gene Ther. 17, 1206–1213 (2010).
Almåsbak, H. et al. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 22, 391–403 (2015).
Bridgeman, J. S. et al. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 184, 6938–6949 (2010).
Guedan, S. et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 3, 1 (2018).
Zhang, T., Wu, M.-R. & Sentman, C. L. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J. Immunol. 189, 2290–2299 (2012).
Dotti, G., Gottschalk, S., Savoldo, B. & Brenner, M. K. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunol. Rev. 257, 107–126 (2014).
Alabanza, L. et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol. Ther. 25, 2452–2465 (2017).
Gross, G., Waks, T. & Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA. 86, 10024–10028 (1989).
Rafiq, S., Hackett, C. S. & Brentjens, R. J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy.Nat. Rev. Clin. Oncol 17, 147–167 (2020).
Brocker, T. & Karjalainen, K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. J. Exp. Med. 181, 1653–1659 (1995).
Till, B. G. et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 112, 2261–2271 (2008).
Hege, K. M. et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J. Immunother. Cancer. 5, 22 (2017).
Brentjens, R. J. et al. Eradication of systemic B-cell tumors by genetically targeted human T lymphocytes co-stimulated by CD80 and interleukin-15. Nat. Med. 9, 279–286 (2003).
Maher, J., Brentjens, R. J., Gunset, G., Rivière, I. & Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat. Biotechnol. 20, 70–75 (2002).
Imai, C. et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 18, 676–684 (2004).
Kawalekar, O. U. et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T Cells. Immunity. 44, 380–390 (2016).
van der Stegen, S. J. C., Hamieh, M. & Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 14, 499–509 (2015).
Guedan, S. et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood. 124, 1070–1080 (2014).
Song, D.-G. & Powell, D. J. Pro-survival signaling via CD27 costimulation drives effective CAR T-cell therapy. Oncoimmunology. 1, 547–549 (2012).
Mata, M. et al. Inducible activation of MyD88 and CD40 in CAR T cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. 7, 1306–1319 (2017).
Hombach, A. A., Heiders, J., Foppe, M., Chmielewski, M. & Abken, H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+ T cells. Oncoimmunology. 1(Jul), 458–466 (2012).
Pulè, M. A. et al. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 12, 933–941 (2005).
Zhong, X.-S., Matsushita, M., Plotkin, J., Riviere, I. & Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 18, 413–420 (2010).
Abate-Daga, D. et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum. Gene Ther. 25, 1003–1012 (2014).
Milone, M. C. et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol. Ther. 17, 1453–1464 (2009).
Majzner, R. G. & Mackall, C. L. Tumor antigen escape from CAR T-cell therapy. Cancer Discov. 8, 1219–1226 (2018).
Maude, S. L., Teachey, D. T., Porter, D. L. & Grupp, S. A. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 125, 4017–4023 (2015).
Green, D. J. et al. Fully human bcma targeted chimeric antigen receptor T cells administered in a defined composition demonstrate potency at low doses in advanced stage high risk multiple myeloma. Blood. 132, 1011–1011 (2018).
Brudno, J. N. et al. T cells genetically modified to express an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of poor-prognosis relapsed multiple myeloma. J. Clin. Oncol. 36, 2267–2280 (2018).
Cohen, A. D. et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Invest. 129, 2210–2221 (2019).
Brown, C. E. et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. of Med. 375, 2561–2569 (2016).
Zhang, H. et al. A Bcma and CD19 bispecific CAR-T for relapsed and refractory multiple myeloma. Blood. 134, 3147–3147 (2019).
Lin, Q., Zhao, J., Song, Y. & Liu, D. Recent updates on CAR T clinical trials for multiple myeloma. Mol. Cancer. 18, 154 (2019).
Dai, H. et al. Bispecific CAR-T cells targeting both CD19 and CD22 for therapy of adults with relapsed or refractory B cell acute lymphoblastic leukemia. J. of Hematol. & Oncol. 13, 30 (2020).
Hossain, N. et al. Phase I experience with a bi-specific CAR targeting CD19 and CD22 in adults with B-cell malignancies. Blood. 132, 490–490 (2018).
Wilkie, S. et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 32, 1059–1070 (2012).
Hegde, M. et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest. 126, 3036–3052 (2016).
Steentoft, C. et al. Glycan-directed CAR-T cells. Glycobiology. 28, 656–669 (2018).
Du, H. et al. Antitumor responses in the absence of toxicity in solid tumors by targeting B7-H3 via chimeric antigen receptor T cells. Cancer Cell. 35, 221–237.e8 (2019).
Majzner, R. G. et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin. Cancer Res. 25, 2560–2574 (2019).
Koneru, M., O’Cearbhaill, R., Pendharkar, S., Spriggs, D. R. & Brentjens, R. J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 13, 102 (2015).
Chekmasova, A. A. et al. Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin. Cancer Res. 16, 3594–3606 (2010).
Murad, J. P. et al. Effective targeting of TAG72+ peritoneal ovarian tumors via regional delivery of CAR-engineered T cells. Front. Immunol. 9, 2268 (2018).
Peter P. Lee (eds.) Tumor Microenvironment (Springer) https://www.springer.com/gp/book/9783030388614
Priceman, S. J. et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2+ breast cancer metastasis to the brain. Clin. Cancer Res. 24, 95–105 (2018).
Brown, C. E. et al. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol. Ther. 26, 31–44 (2018).
Adusumilli, P. S. et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 6, 261ra151 (2014).
Whilding, LM. et al. CAR T-cells targeting the integrin αvβ6 and co-expressing the chemokine receptor cxcr2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers (Basel) 11, 674 (2019).
Liu, G. et al. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 50, 712–724 (2020).
Jin, L. et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors.Nat. Commun. 10, 4016 (2019).
Zhang, B.-L. et al. Hurdles of CAR-T cell-based cancer immunotherapy directed against solid tumors. Sci. China Life Sci. 59, 340–348 (2016).
Caruana, I. et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 21, 524–529 (2015).
Wang, L.-C. S. et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor t cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2, 154–166 (2014).
Quail, D. F. & Joyce, J. A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 19, 1423–1437 (2013).
Yin, Y. et al. Checkpoint blockade reverses anergy in IL-13Rα2 Humanized scFv-Based CAR T cells to treat murine and canine gliomas. Mol. Ther. Oncolytics. 11, 20–38 (2018).
Grosser, R., Cherkassky, L., Chintala, N. & Adusumilli, P. S. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell. 36, 471–482 (2019).
Li, A. M. et al. Checkpoint inhibitors augment CD19-directed chimeric antigen receptor (CAR) T cell therapy in relapsed B-cell acute lymphoblastic leukemia. Blood. 132, 556–556 (2018).
Chong, E. A. et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood. 129, 1039–1041 (2017).
Adusumilli, P. S. et al. Abstract CT036: a phase I clinical trial of malignant pleural disease treated with regionally delivered autologous mesothelin-targeted CAR T cells: safety and efficacy. Cancer Res. 79, CT036–CT036 (2019).
Kloss, C. C. et al. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 26, 1855–1866 (2018).
Chmielewski, M., Hombach, A. A. & Abken, H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol. Rev. 257, 83–90 (2014).
Koneru, M., Purdon, T. J., Spriggs, D., Koneru, S. & Brentjens, R. J. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 4, e994446 (2015).
Krenciute, G. et al. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol. Res. 5, 571–581 (2017).
Mohammed, S. et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol. Ther. 25, 249–258 (2017).
Roex, G. et al. Safety and clinical efficacy of BCMA CAR-T-cell therapy in multiple myeloma. J. of Hematol. & Oncol. 13, 164 (2020).
Research C for DE and. FDA approves tisagenlecleucel for B-cell ALL and tocilizumab for cytokine release syndrome. FDA https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-tisagenlecleucel-b-cell-all-and-tocilizumab-cytokine-release-syndrome.
Research C for DE and. FDA approves axicabtagene ciloleucel for large B-cell lymphoma. FDA https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-axicabtagene-ciloleucel-large-b-cell-lymphoma.
Park, J. H. et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. of Med. 378, 449–459 (2018).
Frey, N. V. & Porter, D. L. Cytokine release syndrome with novel therapeutics for acute lymphoblastic leukemia. Hematol. Am. Soc. Hematol. Educ. Program. 2016, 567–572 (2016).
Santomasso, B. D. et al. Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with b-cell acute lymphoblastic leukemia. Cancer Discov. 8, 958–971 (2018).
Sterner, R. & Kenderian, S. Myeloid cell and cytokine interactions with chimeric antigen receptor-T-cell therapy: implication for future therapies. Curr. Opin. in Hematol. 27, 41–48 (2020).
Neelapu, S. S. et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat. Rev. Clin. Oncol. 15, 47–62 (2018).
Lee, D. W. et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol. Blood Marrow Transplant. 25, 625–638 (2019).
Davila, M. L. et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 6, 224ra25 (2014).
Lee, D. W. et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 124, 188–195 (2014).
Milone, M. C. & Bhoj, V. G. The pharmacology of T cell therapies. Mol. Ther. Methods Clin. Dev. 8, 210–221 (2018).
Ying, Z. et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 25, 947–953 (2019).
Salter, AI. et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci. Signal 11, 544 (2018).
Sommermeyer, D. et al. Fully human CD19-specific chimeric antigen receptors for T-cell therapy. Leukemia. 31, 2191–2199 (2017).
Jonnalagadda, M. et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid Fc receptor binding and improve t cell persistence and antitumor efficacy. Mol. Ther. 23, 757–768 (2015).
Locke, F. L. et al. Preliminary results of prophylactic tocilizumab after axicabtageneciloleucel (axi-cel; KTE-C19) treatment for patients with refractory,aggressive non-hodgkin lymphoma (NHL). Blood. 130, 1547–1547 (2017).
Sterner, R. M. et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 133, 697–709 (2019).
Sterner, RM., Cox, MJ., Sakemura, R. & Kenderian, SS. Using CRISPR/Cas9 to Knock Out GM-CSF in CAR-T Cells. J. Vis. Exp. 22, 149 (2019).
Sachdeva, M., Duchateau, P., Depil, S., Poirot, L. & Valton, J. Granulocyte-macrophage colony-stimulating factor inactivation in CAR T-cells prevents monocyte-dependent release of key cytokine release syndrome mediators. J. Biol. Chem. 294, 5430–5437 (2019).
Staedtke, V. et al. Disruption of a self-amplifying catecholamine loop reduces cytokine release syndrome. Nature. 564, 273–277 (2018).
Giavridis, T. et al. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 24, 731–738 (2018).
Jones, B. S., Lamb, L. S., Goldman, F., Di & Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 5, 254 (2014).
Philip, B. et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood. 124, 1277–1287 (2014).
Di Stasi, A. et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 365, 1673–1683 (2011).
Juillerat, A. et al. Modulation of chimeric antigen receptor surface expression by a small molecule switch. BMC Biotechnol. 19, 44 (2019).
Mestermann K. et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med. (2019). https://stm.sciencemag.org/content/11/499/eaau5907.
Acknowledgements
This work was supported through Regenerative Medicine Minnesota RMM 012819 EPC 003 (RMS). RCS is a student in the University of Wisconsin-Madison Medical Scientist Training Program supported by T32GM008692.
Author information
Authors and Affiliations
Contributions
R.C.S. and R.M.S. designed, wrote, edited, and approved the final version of the manuscript. R.M.S. supervised the study.
Corresponding author
Ethics declarations
Conflict of interest
R.M.S. is an inventor on patents related to CAR-T cell therapy licensed to Humanigen through Mayo Clinic.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sterner, R.C., Sterner, R.M. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 11, 69 (2021). https://doi.org/10.1038/s41408-021-00459-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41408-021-00459-7
This article is cited by
-
Melatonin as an immunomodulator in CD19-targeting CAR-T cell therapy: managing cytokine release syndrome
Journal of Translational Medicine (2024)
-
Myeloid leukemia-derived galectin-1 downregulates CAR expression to hinder cytotoxicity of CAR T cells
Journal of Translational Medicine (2024)
-
New immunotherapy approaches for colorectal cancer: focusing on CAR-T cell, BiTE, and oncolytic viruses
Cell Communication and Signaling (2024)
-
Current challenges and therapeutic advances of CAR-T cell therapy for solid tumors
Cancer Cell International (2024)
-
Adoptive cell transfer therapy with ex vivo primed peripheral lymphocytes in combination with anti-PDL1 therapy effectively inhibits triple-negative breast cancer growth and metastasis
Molecular Cancer (2024)
