
Abstract
Extraskeletal myxoid chondrosarcoma is a rare subtype of sarcoma that affects the soft tissue and bones in middle-aged and elderly adults. Its diagnosis can be challenging, with the differential diagnoses including a wide variety of mesenchymal tumors. The line of differentiation of extraskeletal myxoid chondrosarcoma has been controversial, but recent evidence suggests a neuroendocrine phenotype. INSM1 is a zinc-finger transcription factor that plays a pivotal role in neuroendocrine differentiation, and has been proposed as a promising immunohistochemical marker of neuroendocrine carcinoma. The aim of this study was to determine the prevalence of INSM1 expression in extraskeletal myxoid chondrosarcoma and to understand its significance in sarcoma diagnosis. We immunostained the representative sections of 31 NR4A3-rearranged extraskeletal myxoid chondrosarcomas and 187 histological mimics. Nuclear staining of moderate or higher intensity in at least 5% of tumor cells was considered positive. Twenty-eight of the 31 extraskeletal myxoid chondrosarcomas (90%) were positive for INSM1, providing strong evidence for neuroendocrine differentiation. The staining was diffuse (>50%) in 17 cases, with most immunopositive tumors showing at least focal strong expression. The INSM1 staining extent was not correlated with cytomorphology, synaptophysin expression, or fusion types (EWSR1 vs non-EWSR1). In contrast, INSM1 expression was negative in 94% of the 187 other mesenchymal tumors. INSM1-positive mimics comprised a small subset of chordoma (1 of 10), soft tissue myoepithelioma (1 of 20), ossifying fibromyxoid tumor (3 of 10), and Ewing sarcoma (3 of 10), among other tumor types. The majority of these cases showed labeling in <25% of the tumor cells. Although not entirely sensitive or specific, INSM1 could be a potential marker for the diagnosis of extraskeletal myxoid chondrosarcoma when molecular genetic access is limited.
Similar content being viewed by others
Main
Extraskeletal myxoid chondrosarcoma is a rare subtype of sarcoma that typically affects the deep soft tissue of adults mainly in the 4th to 7th decade of life, with an approximately 2:1 male predominance.1 Rare examples of primary tumors in the bone are well documented.2 Histologically, extraskeletal myxoid chondrosarcoma is a multinodular growth populated by mildly cohesive spindle to epithelioid cells that are arranged in cords, strands, pseudoacini, or clusters within abundant myxoid stroma. The tumor cells harbor uniform nuclei and deeply eosinophilic cytoplasm. Histological variations include a cellular variant that may exhibit inconspicuous mucin and rhabdoid cytology.3, 4 Extraskeletal myxoid chondrosarcoma harbors specific recurrent chromosomal translocations in over 95% of cases, which create fusions involving the whole coding sequence of NR4A3 (NOR1, CHN, or TEC, located at 9q22). The most frequent fusion partner of NR4A3 is EWSR1 (22q12, 60–75%),5 followed by TAF15 (RBP56 or TAF2N; 17q11, 15–20%), and other rare targets, such as TCF12 (15q21), TFG (3q12),6 FUS (16p11),7 and HSPA8 (11q24.1),8 each accounting for <3%.9 Surgical resection is the primary therapy and the tumor follows a relatively protracted clinical course with a propensity for late recurrence and distant metastases. According to multivariate analysis, older age, tumor size, and proximal location are associated with poor survival, while histological factors did not contribute significantly to tumor behavior.10 However, some histological (eg, cellularity, mitotic activity, and atypia) and molecular (eg, EWSR1 vs non-EWSR1 fusion partner) parameters may predict behavior according to univariate analyses.3, 11
The lineage of extraskeletal myxoid condrosarcoma has been controversial. Many of the tumors reported by the name of chordoid sarcoma or soft-tissue chondrosarcoma12 in the literature were re-categorized into extraskeletal myxoid chondrosarcoma, when Enzinger and Shiraki proposed this concept as a distinct form of chondrosarcoma.13 The chondroblastic nature of this sarcoma was then believed to be supported by chondroitin-4 and 6-sulphate-rich extracellular mucin, S100 protein expression, and ultrastructural resemblance to chondroblasts.14 However, these findings have become considered insufficiently specific for cartilaginous differentiation, and indeed, the extracellular matrix of extraskeletal myxoid chondrosarcoma mostly lack type II and X collagen, with only exceptional true hyaline cartilage formation.15 Recent investigations have instead suggested neuroendocrine/neural differentiation.3, 14, 16 Ultrastructurally, membrane-bound dense core granules, compatible with neurosecretory granules, were present in some cases.14, 16 Immunohistochemically, extraskeletal myxoid chondrosarcomas are variably positive for synaptophysin with a range of 22–87%.3, 14, 17, 18 Other positive markers that are suggestive of neuroendocrine/neural differentiation include: chromogranin A (0–4%),3, 14, 16, 17 PGP9.5 (7–40%),14, 17 microtubule-associated protein-2 (84%),19 class III β-tubulin (52%),19 and peripherin (53–60%).17, 20 However, some of these markers are not exclusively specific for a neuroendocrine/neural lineage. The current WHO classification does not consider extraskeletal myxoid chondrosarcoma as of cartilaginous or neural lineage, but rather classify this malignancy under the section of tumors of uncertain differentiation.1
A diagnosis of extraskeletal myxoid chondrosarcoma can be challenging. The histological spectrum is wide, and needle biopsy, which is becoming increasingly utilized, may not capture characteristic patterns. In addition, the latest classification scheme of soft-tissue tumors has recognized several newly characterized histological mimics, such as myoepithelial tumors and CIC-rearranged sarcomas. Differential diagnoses include myxoid neoplasms and some round-cell tumors, both benign and malignant, with accurate diagnosis being crucial for appropriate management. Molecular detection of pathognomonic fusion genes involving NR4A3 is probably the most robust method for diagnosis.5, 9, 11, 21, 22 However, this approach requires cost, labor, and expertise, and is only available at large medical centers; therefore, immunohistochemistry is expected to be a practical surrogate. S100 protein has been a conventional marker since the era when this sarcoma was believed to be a form of chondrosarcoma; however, it has a notably poor sensitivity (13–50%),3, 11, 14, 17, 18 low specificity, and often only stains focally, with weak intensity even when it is positive.3, 11, 14, 17 Synaptophysin staining is considered characteristic, but the reported frequency of reactivity varies greatly (22–87%) and its specificity has not been systematically tested.
INSM1 is a zinc-finger transcription factor that was first identified using a human insulinoma tissue subtraction library.23 INSM1 plays a pivotal role in the development and differentiation of pancreatic and gastrointestinal neuroendocrine cells, adrenal medulla, and neuronal progenitor cells.24, 25, 26, 27 The expression of INSM1 is abundant in the developing neuroendocrine/neural system, while it is restricted in normal adult tissues.28 Immunohistochemical studies using a monoclonal antibody to INSM1, have shown that most neuroendocrine cells are positively labeled, such as those in the gastrointestinal, pancreatic, bronchopulmonary, and adrenal medullary tissues, while non-neuroendocrine cells are negative.29, 30 INSM1 expression has also been tested in human neoplasms, with expression in 93–98% of non-parathyroidal neuroendocrine/neuroepithelial neoplasms of both low and high grades.29, 30 INSM1 has been specifically tested in the context of thoracic and gynecologic pathology,31, 32, 33 and is emerging as a highly sensitive and specific neuroendocrine marker that could outperform the conventional triad of synaptophysin, chromogranin A, and CD56.33 INSM1 expression has never been tested in sarcomas, except two cases of primitive neuroectodermal tumors.29 Due to the potential neuroendocrine/neural differentiation in extraskeletal myxoid chondrosarcoma and the essential role of INSM1 in such a differentiation, we aimed to determine (1) whether INSM1 is expressed in extraskeletal myxoid chondrosarcoma, and (2) if its detection could be helpful in diagnosis.
Materials and methods
Cases
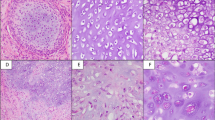
After the approval by the institutional review board (2014−089), 31 cases of extraskeletal myxoid chondrosarcoma were retrieved from the pathology files of the National Cancer Center Hospital, Tokyo, Japan. The diagnoses of all cases were confirmed by morphological examination, conventional immunohistochemistry, and molecular assays. All 31 tumors were previously confirmed to harbor NR4A3 rearrangement by fluorescence in situ hybridization (FISH). In addition, all cases were previously tested for EWSR1 rearrangement status by FISH, and 24 tumors (77%) were positive for EWSR1 rearrangement, while 7 tumors (23%) were negative. Extraskeletal myxoid chondrosarcoma occurred in 19 men and 12 women with an age range of 32–86 years (median, 54 years old). Two tumors primarily developed in the bone (the proximal phalanx of the great toe and the sacrum), and the remaining tumors arose from the soft tissue. Histologically, 29 tumors showed at least focal conventional myxoid morphology, while 2 tumors were entirely composed of round cells without intervening myxoid stroma. The predominant tumor cytology was long spindle in 4, short spindle in 17, and round in 10 (Figure 1). Rhabdoid cells were present in 6 cases.

Extraskeletal myxoid chondrosarcoma demonstrated a spectrum of histomorphology. The predominant tumor cytology was short spindle (a), long spindle (b), or round (c). A purely cellular variant showed densely packed round cells with colloid-like secretion, without myxoid stroma (d).
We also selected 187 mesenchymal tumors that were thought to mimic extraskeletal myxoid chondrosarcoma. These included 20 skeletal chondrosarcomas, 10 chordomas, 20 myxofibrosarcomas, 20 myxoid liposarcomas, 15 intramuscular myxomas, 10 low-grade fibromyxoid sarcomas, 20 soft-tissue myoepitheliomas, 10 ossifying fibromyxoid tumors, 5 myxoid variant of angiomatoid fibrous histiocytomas, 10 dedifferentiated liposarcomas with myxoid change, 10 Ewing sarcomas, 5 CIC-rearranged sarcomas, 5 poorly-differentiated synovial sarcomas, 5 BCOR-CCNB3 sarcomas, and 22 miscellaneous tumors (4 malignant rhabdoid tumors, 3 epithelioid malignant peripheral nerve sheath tumors, 2 myxoid solitary fibrous tumors, 2 desmoplastic small round cell tumors, 2 alveolar rhabdomyosarcomas, 2 soft tissue chondroma, 1 chondromyxoid fibroma, 1 acral fibromyxoma, 1 aggressive angiomyxoma, 1 nodular fasciitis, 1 epithelioid sarcoma with myxoid change, 1 superficial angiomyxoma, and 1 lipoblastoma-like tumor of the vulva). In addition, INSM1 expression was evaluated in selected adult human non-neoplastic tissues that were newly prepared from unrelated cases or were incidentally included in the tumor slides.
Immunohistochemistry
Four-micrometer-thick sections from the representative block of each tumor were routinely deparaffinized. The sections were exposed to 3% hydrogen peroxide for 15 min to block endogenous peroxidase activity. Antigen retrieval was performed using water bath (98 °C, 40 min) in Targeted Retrieval Solution pH 9 (Dako, Glostrup, Denmark). The primary antibody used was INSM1 (clone A-8, dilution 1:100, Santa Cruz Biotechnology, Dallas, TX, USA). The slides were incubated for 1 h at room temperature with the primary antibody and subsequently labeled using the EnVision system (Dako). Diaminobenzidine was used as the chromogen and hematoxylin as the counterstain. The intensity of nuclear staining was semi-quantitatively graded as none, weak, moderate, or strong. Weak staining was not visible on a low-power view (× 4 objective), while moderate and strong staining was readily visible on low-power magnification. For evaluation, moderate or higher staining in at least 5% of tumor cells was considered positive, and any reactivity under this level was regarded as negative. The extent of nuclear staining was classified as negative (0 to <5%), 5–25%, 26–50%, 51–75%, and 76–100%. For extraskeletal myxoid chondrosarcoma cases, other immunohistochemical results obtained at the original diagnosis were also recorded.
Results
The immunohistochemical results are summarized in Table 1. Of the 31 extraskeletal myxoid chondrosarcomas, 28 (90%) were positive for INSM1 (Figure 2). The reactivity ranged from 5–100%; it was diffuse (>50%) in 17 cases and extensive (>75%) in 12 cases. Positive staining was of moderate or higher intensity by definition; most (23 of 28) immunopositive tumors showed at least focal strong INSM1 expression, while the remaining 5 tumors showed moderate staining intensity. The INSM1 staining was not significantly correlated with cytomorphology, fusion type (EWSR1 vs non-EWSR1), or disease-specific survival. Three extraskeletal myxoid chondrosarcomas were negative for INSM1, including 1 case of cellular variant. Among INSM1-negative cases, EWSR1 rearrangement was present in 2 cases and absent in 1 case.

Most of the 31 extraskeletal myxoid chondrosarcomas (90%) were immunoreactive to INSM1. The staining varied in range, and can be diffuse (a, b) or focal (c, d).
INSM1 was negative in 176 out of 187 other mesenchymal tumors (94%, Figure 3) with 6% tested being immunoreactive (Figure 4). INSM1-positive mimics comprised of chordoma (1 of 10), soft tissue myoepithelioma (1 of 20), ossifying fibromyxoid tumor (3 of 10), Ewing sarcoma (3 of 10), BCOR-CCNB3 sarcoma (1 of 5), alveolar rhabdomyosarcoma (1 of 2), and epithelioid malignant peripheral nerve sheath tumor (1 of 3). Among the INSM1-positive non-extraskeletal myxoid chondrosarcoma tumors, diffuse (>50%) reactivity was observed in 1 BCOR-CCNB3 sarcoma, while the majority (9 of 11) showed labeling in less than 25% of analyzed cells. At least focal strong staining intensity was observed in 5 mimics (1 ossifying fibromyxoid tumor, 1 chordoma, 2 Ewing sarcoma, and 1 epithelioid malignant peripheral nerve sheath tumor).

Most of the 187 non-extraskeletal myxoid chondrosarcoma mesenchymal tumors (94%) lacked INSM1 reactivity (a), skeletal chondrosarcoma; (b), myxoid liposarcoma; (c), CIC-rearranged sarcoma; (d), myoepithelial tumor).

A small number of non-extraskeletal myxoid chondrosarcoma mesenchymal tumors were positive for INSM1 (a), ossifying fibromyxoid tumor; (b), Ewing sarcoma).
Among the conventional markers evaluated at the original diagnosis of extraskeletal myxoid chondrosarcoma, S100 protein was focally expressed in 15 of 29 tumors (52%) tested. Synaptophysin was positive in 13 of 31 cases (42%) tested, ranging from weak expression in <1% of cells to strong expression in >90% of cells. When synaptophysin positivity was defined as staining in at least 5% of cells, the sensitivity dropped to 26%. Synaptophysin expression was not significantly correlated with the extent of INSM1 reactivity. AE1/AE3, epithelial membrane antigen, smooth muscle actin, desmin, and CD34 were expressed in 0 of 28 (0%), 8 of 25 (32%), 1 of 15 (7%), 0 of 14 (0%), 5 of 24 (21%) cases originally tested.
In adult non-neoplastic tissues, INSM1 expression was tightly restricted to the isolated neuroendocrine cells in the gastrointestinal and bronchial/bronchiolar mucosa, pancreatic islet cells, adrenal medulla, paraganglia, thyroidal C-cells, pituitary gland, and rare neurons in the spinal cord. One unexpected finding was the mostly weak INSM1 reactivity of some dendritic cells in the lymph nodes. Notably, we did not observe INSM1-expressing cells in the cerebrum, cerebellum, or parathyroid. We also did not detect INSM1-positive cells in the subcutis, muscle, or bone, where EMCs are most commonly found.
Discussion
Several lines of evidence have suggested a neuroendocrine/neural differentiation in extraskeletal myxoid chondrosarcoma; however, the variability of immunophenotypes reported in previous studies, as well as the questionable specificity of investigated markers, have hampered firmly establishing their line of differentiation. In this study, we have shown that INSM1, a critical transcription factor for neuroendocrine differentiation, was expressed in 90% of the 31 genetically confirmed extraskeletal myxoid chondrosarcomas. Restricted INSM1 expression in adult non-neoplastic tissues supported the specificity of this staining, and the present data add evidence of a neuroendocrine differentiation in this sarcoma. Interestingly, however, the extent of INSM1 staining was not significantly correlated with synaptophysin expression status. Extraskeletal myxoid chondrosarcoma is also reportedly only rarely positive for chromogranin A.3, 14, 17 Although INSM1 has been reported as an upstream regulator of neuroendocrine effector molecules,31 the neuroendocrine molecular pathway may be functioning in an aberrant manner in this sarcoma type.
Three extraskeletal myxoid chondrosarcomas (10%) were nonreactive to INSM1. One of these resection specimens was previously decalcified by acid. In another case, repeat staining on a preoperative biopsy demonstrated focal INSM1 reactivity, which may suggest some contribution of pre-analytic factors for immunoreactivity. Extraskeletal myxoid chondrosarcoma is often large, highly myxoid, and hemorrhagic, and is therefore likely to be fixed for an extended period to facilitate grossing. Although INSM1 staining was only focal (≤50%) in 39% of the positive cases, how this may compromise the value of staining in biopsy interpretation remains unclear. This is because biopsies are more likely to be fixed appropriately and some tumors that were focally positive for INSM1 showed interspersed reactivity, which could be readily detected in small specimens. Our cohort included three needle biopsy specimens, with all showing positivity for INSM1. In this regard, one drawback of INSM1 staining is the lack of internal positive control in most tumor sections, as its expression is limited in normal adult tissues.
INSM1 expression was absent in most other mesenchymal mimics tested, which suggests its potential as a diagnostic biomarker. However, the staining was not completely specific, and it is particularly concerning that INSM1 was expressed in a minor subset of tumor entities that most aptly mimic extraskeletal myxoid chondrosarcoma. These included 5% of myoepithelial tumors and 30% of ossifying fibromyxoid tumors, with NR4A3 testing potentially being required in some of these cases. INSM1 expression in these tumors were nonetheless limited in extent, and only one of the tumors showed diffuse (>50%) reactivity. The only INSM1-positive myoepithelial tumor in our cohort was a non-myxoid malignant myoepithelioma, which diffusely expressed cytokeratin and showed moderate INSM1 expression in 10% of the cells. None of the extraskeletal myxoid chondrosarcomas studied expressed cytokeratin, and previous studies reported an exceptionally rare occurrence of keratin expression,3, 5, 14, 17 unlike >90% of myoepithelial tumors.34 Many ossifying fibromyxoid tumors demonstrate mature bone formation. In addition, their often fibromyxoid to sometimes hyalinized quality of the stroma is different from purely myxoid stromal properties of extraskeletal myxoid chondrosarcoma. Ossifying fibromyxoid tumors express desmin in about 40% of cases, which is an incompatible phenotype with extraskeletal myxoid chondrosarcoma. INSM1 expression in some ossifying fibromyxoid tumors may be more than incidental, as there is some gene expression data suggesting a neural/neuroendocrine differentiation.35
The mechanism of INSM1 upregulation in extraskeletal myxoid chondrosarcomas is unclear. The pathogenomonic fusion protein of these sarcomas consists of the complete amino-acid sequence of NR4A3 with the DNA-binding domain.36 Transfection experiments showed that EWSR1-NR4A3 is a highly potent transcriptional activator,37 suggesting its oncogenic role by activating specific genes. However, the actual downstream target of fusion and the detailed mechanism of oncogenesis are not completely understood. A few targets that were previously suggested include peroxisome proliferator-activated receptor gamma (PPARG)38 and serum- and glucocorticoid-regulated kinase 1 (SGK1),39 with our present data possibly suggesting INSM1 as another target. However, in a study using human bone marrow mesenchymal cell lines transfected with an EWSR1-NR4A3 expression vector, INSM1 was not included in the 317 significantly upregulated genes.40
INSM1 gene expression in extraskeletal myxoid chondrosarcoma has been documented in a few previous expression microarray analyses, but it has never been expected to be a promising biomarker. In Subramanian et al,41 INSM1 was differentially upregulated in 10 extraskeletal myxoid chondrosarcomas compared with 5 other types of sarcomas (ie, dermatofibrosarcoma protuberans, synovial sarcoma, leiomyosarcomas, malignant fibrous histiocytoma, and gastrointestinal stromal tumor), but this was not included among the top 112 genes in the significance analysis of microarrays. In the paper by Filion et al,38 using three extraskeletal myxoid chondrosarcomas and five other types of sarcomas (ie, Ewing sarcoma, alveolar rhabdomyosarcoma, desmoplastic small round cell tumor, alveolar soft part sarcoma, and synovial sarcoma), INSM1 was differentially expressed in extraskeletal myxoid chondrosarcomas but this was not statistically significant after statistical adjustment. In a study by Sjögren et al,42 2 extraskeletal myxoid chondrosarcomas and a single myxoid liposarcoma were compared, with INSM1 not being included in the top 35 genes that were differentially expressed. This apparent discordance between our protein-based results and published gene expression data may be related to post transcriptional modulation, antibody characteristics, antibody availability, and the study context. Regarding the latter, in neither of the first two large studies were the control sarcoma types selected on the basis of histologic context, and most mimics included in the present study were not tested.
In conclusion, INSM1 is frequently expressed in extraskeletal myxoid chondrosarcoma, which adds evidence to the suspected neuroendocrine/neural differentiation. INSM1 was also found to be a potential diagnostic biomarker for this tumor. A small fraction of extraskeletal myxoid chondrosarcoma was negative for INSM1 and a small number of other mesenchymal tumors were found to be positive. Nonetheless, diffuse and at least moderate INSM1 positivity seems specific for extraskeletal myxoid chondrosarcoma in an appropriate histologic context. The present study expands the utility of INSM1 staining beyond neuroendocrine carcinomas and suggests its significance in sarcoma diagnosis when molecular genetic access is limited.
References
Lucas DR, Stenman G. Extraskeletal myxoid chondrosarcoma. In: Fletcher CDM, Bridge JA, Hogendoorn PCW et al(eds). WHO classification of tumours of soft tissue and bone. IARC Press: Lyon, 2013, pp 223–224.
Demicco EG, Wang WL, Madewell JE et al. Osseous myxochondroid sarcoma: a detailed study of 5 cases of extraskeletal myxoid chondrosarcoma of the bone. Am J Surg Pathol 2013;37:752–762.
Oliveira AM, Sebo TJ, McGrory JE et al. Extraskeletal myxoid chondrosarcoma: a clinicopathologic, immunohistochemical, and ploidy analysis of 23 cases. Mod Pathol 2000;13:900–908.
Lucas DR, Fletcher CD, Adsay NV et al. High-grade extraskeletal myxoid chondrosarcoma: a high-grade epithelioid malignancy. Histopathology 1999;35:201–208.
Antonescu CR, Argani P, Erlandson RA et al. Skeletal and extraskeletal myxoid chondrosarcoma: a comparative clinicopathologic, ultrastructural, and molecular study. Cancer 1998;83:1504–1521.
Hisaoka M, Ishida T, Imamura T et al. TFG is a novel fusion partner of NOR1 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 2004;40:325–328.
Broehm CJ, Wu J, Gullapalli RR et al. Extraskeletal myxoid chondrosarcoma with a t(9;16)(q22;p11.2) resulting in a NR4A3-FUS fusion. Cancer Genet 2014;207:276–280.
Urbini M, Astolfi A, Pantaleo MA et al. HSPA8 as a novel fusion partner of NR4A3 in extraskeletal myxoid chondrosarcoma. Genes Chromosomes Cancer 2017;56:582–586.
Benini S, Cocchi S, Gamberi G et al. Diagnostic utility of molecular investigation in extraskeletal myxoid chondrosarcoma. J Mol Diagn 2014;16:314–323.
Meis-Kindblom JM, Bergh P, Gunterberg B et al. Extraskeletal myxoid chondrosarcoma: a reappraisal of its morphologic spectrum and prognostic factors based on 117 cases. Am J Surg Pathol 1999;23:636–650.
Agaram NP, Zhang L, Sung YS et al. Extraskeletal myxoid chondrosarcoma with non-EWSR1-NR4A3 variant fusions correlate with rhabdoid phenotype and high-grade morphology. Hum Pathol 2014;45:1084–1091.
Stout AP, Verner EW . Chondrosarcoma of the extraskeletal soft tissues. Cancer 1953;6:581–590.
Enzinger FM, Shiraki M . Extraskeletal myxoid chondrosarcoma. An analysis of 34 cases. Hum Pathol 1972;3:421–435.
Goh YW, Spagnolo DV, Platten M et al. Extraskeletal myxoid chondrosarcoma: a light microscopic, immunohistochemical, ultrastructural and immuno-ultrastructural study indicating neuroendocrine differentiation. Histopathology 2001;39:514–524.
Aigner T, Oliveira AM, Nascimento AG . Extraskeletal myxoid chondrosarcomas do not show a chondrocytic phenotype. Mod Pathol 2004;17:214–221.
Harris M, Coyne J, Tariq M et al. Extraskeletal myxoid chondrosarcoma with neuroendocrine differentiation: a pathologic, cytogenetic, and molecular study of a case with a novel translocation t(9;17)(q22;q11.2). Am J Surg Pathol 2000;24:1020–1026.
Okamoto S, Hisaoka M, Ishida T et al. Extraskeletal myxoid chondrosarcoma: a clinicopathologic, immunohistochemical, and molecular analysis of 18 cases. Hum Pathol 2001;32:1116–1124.
Shao R, Lao IW, Wang L et al. Clinicopathologic and radiologic features of extraskeletal myxoid chondrosarcoma: a retrospective study of 40 Chinese cases with literature review. Ann Diagn Pathol 2016;23:14–20.
Hisaoka M, Okamoto S, Koyama S et al. Microtubule-associated protein-2 and class III beta-tubulin are expressed in extraskeletal myxoid chondrosarcoma. Mod Pathol 2003;16:453–459.
Cummings TJ, Shea CR, Reed JA et al. Expression of the intermediate filament peripherin in extraskeletal myxoid chondrosarcoma. J Cutan Pathol 2000;27:141–146.
Flucke U, Tops BB, Verdijk MA et al. NR4A3 rearrangement reliably distinguishes between the clinicopathologically overlapping entities myoepithelial carcinoma of soft tissue and cellular extraskeletal myxoid chondrosarcoma. Virchows Arch 2012;460:621–628.
Noguchi H, Mitsuhashi T, Seki K et al. Fluorescence in situ hybridization analysis of extraskeletal myxoid chondrosarcomas using EWSR1 and NR4A3 probes. Hum Pathol 2010;41:336–342.
Goto Y, De Silva MG, Toscani A et al. A novel human insulinoma-associated cDNA, IA-1, encodes a protein with "zinc-finger" DNA-binding motifs. J Biol Chem 1992;267:15252–15257.
Farkas LM, Haffner C, Giger T et al. Insulinoma-associated 1 has a panneurogenic role and promotes the generation and expansion of basal progenitors in the developing mouse neocortex. Neuron 2008;60:40–55.
Rosenbaum JN, Duggan A, Garcia-Anoveros J . Insm1 promotes the transition of olfactory progenitors from apical and proliferative to basal, terminally dividing and neuronogenic. Neural Dev 2011;6:6.
Wildner H, Gierl MS, Strehle M et al. Insm1 (IA-1) is a crucial component of the transcriptional network that controls differentiation of the sympatho-adrenal lineage. Development 2008;135:473–481.
Gierl MS, Karoulias N, Wende H et al. The zinc-finger factor Insm1 (IA-1) is essential for the development of pancreatic beta cells and intestinal endocrine cells. Genes Dev 2006;20:2465–2478.
Xie J, Cai T, Zhang H et al. The zinc-finger transcription factor INSM1 is expressed during embryo development and interacts with the Cbl-associated protein. Genomics 2002;80:54–61.
Rosenbaum JN, Guo Z, Baus RM et al. INSM1: a novel immunohistochemical and molecular marker for neuroendocrine and neuroepithelial neoplasms. Am J Clin Pathol 2015;144:579–591.
Fujino K, Yasufuku K, Kudoh S et al. INSM1 is the best marker for the diagnosis of neuroendocrine tumors: comparison with CGA, SYP and CD56. Int J Clin Exp Pathol 2017;10:5393–5405.
Fujino K, Motooka Y, Hassan WA et al. Insulinoma-associated protein 1 is a crucial regulator of neuroendocrine differentiation in lung cancer. Am J Pathol 2015;185:3164–3177.
Kuji S, Watanabe R, Sato Y et al. A new marker, insulinoma-associated protein 1 (INSM1), for high-grade neuroendocrine carcinoma of the uterine cervix: Analysis of 37 cases. Gynecol Oncol 2017;144:384–390.
Rooper LM, Sharma R, Li QK et al. INSM1 demonstrates superior performance to the individual and combined use of synaptophysin, chromogranin and CD56 for diagnosing neuroendocrine tumors of the thoracic cavity. Am J Surg Pathol 2017;41:1561–1569.
Hornick JL, Fletcher CD . Myoepithelial tumors of soft tissue: a clinicopathologic and immunohistochemical study of 101 cases with evaluation of prognostic parameters. Am J Surg Pathol 2003;27:1183–1196.
Graham RP, Dry S, Li X et al. Ossifying fibromyxoid tumor of soft parts: a clinicopathologic, proteomic, and genomic study. Am J Surg Pathol 2011;35:1615–1625.
Labelle Y, Zucman J, Stenman G et al. Oncogenic conversion of a novel orphan nuclear receptor by chromosome translocation. Hum Mol Genet 1995;4:2219–2226.
Labelle Y, Bussieres J, Courjal F et al. The EWS/TEC fusion protein encoded by the t(9;22) chromosomal translocation in human chondrosarcomas is a highly potent transcriptional activator. Oncogene 1999;18:3303–3308.
Filion C, Motoi T, Olshen AB et al. The EWSR1/NR4A3 fusion protein of extraskeletal myxoid chondrosarcoma activates the PPARG nuclear receptor gene. J Pathol 2009;217:83–93.
Poulin H, Filion C, Ladanyi M et al. Serum- and glucocorticoid-regulated kinase 1 (SGK1) induction by the EWS/NOR1(NR4A3) fusion protein. Biochem Biophys Res Commun 2006;346:306–313.
Filion C, Labelle Y . Identification of genes regulated by the EWS/NR4A3 fusion protein in extraskeletal myxoid chondrosarcoma. Tumour Biol 2012;33:1599–1605.
Subramanian S, West RB, Marinelli RJ et al. The gene expression profile of extraskeletal myxoid chondrosarcoma. J Pathol 2005;206:433–444.
Sjögren H, Meis-Kindblom JM, Orndal C et al. Studies on the molecular pathogenesis of extraskeletal myxoid chondrosarcoma-cytogenetic, molecular genetic, and cDNA microarray analyses. Am J Pathol 2003;162:781–792.
Acknowledgements
We thank Sachiko Miura, Toshiko Sakaguchi, and Chizu Kina for technical assistance. The study was supported in part by JSPS Grant-in-Aid for Young Scientists (15K19065).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yoshida, A., Makise, N., Wakai, S. et al. INSM1 expression and its diagnostic significance in extraskeletal myxoid chondrosarcoma. Mod Pathol 31, 744–752 (2018). https://doi.org/10.1038/modpathol.2017.189
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2017.189
This article is cited by
-
Extraskeletal myxoid chondrosarcoma: p53 and Ki-67 offer prognostic value for clinical outcome — an immunohistochemical and molecular analysis of 31 cases
Virchows Archiv (2023)
-
Mutation of KIT in cellular extraskeletal myxoid chondrosarcoma: a case report and literature review
Diagnostic Pathology (2022)
-
Lung neuroendocrine neoplasms: recent progress and persistent challenges
Modern Pathology (2022)
-
Update from the 5th Edition of the World Health Organization Classification of Head and Neck Tumors: Soft Tissue Tumors
Head and Neck Pathology (2022)
-
Primary small intestine mesenteric low-grade fibromyxoid sarcoma with foci of atypical epithelioid whorls and diffuse DOG1 expression: a case report
Diagnostic Pathology (2020)
