
Abstract
Detection of minimal residual disease predicts adverse outcome in patients with acute myeloid leukemia. Currently, minimal residual disease may be detected by RQ-PCR or flow cytometry, both of which have practical and diagnostic limitations. Here, we describe a next-generation sequencing assay for minimal residual disease detection in NPM1-mutated acute myeloid leukemia, which encompasses ∼60% of patients with normal karyotype acute myeloid leukemia. Exon 12 of NPM1 was PCR amplified using sequencing adaptor-linked primers and deep sequenced to enable detection of low-prevalence, acute myeloid leukemia-specific activating mutations. We benchmarked our results against flow cytometry, the standard of care for acute myeloid leukemia minimal residual disease diagnosis at our institution. The performance of both approaches was evaluated using defined dilutions of an NPM1 mutation-positive cell line and longitudinal clinical samples from acute myeloid leukemia patients. Using defined control material, we found this assay sensitive to approximately 0.001% mutant cells, outperforming flow cytometry by an order of magnitude. Next-generation sequencing was precise and semiquantitative over four orders of magnitude. In 22 longitudinal samples from six acute myeloid leukemia patients, next-generation sequencing detected minimal residual disease in all samples deemed negative by flow cytometry. Further, in one-third of patients, sequencing detected alternate NPM1 mutations in addition to the patient’s index mutation, consistent with tumor heterogeneity. Next-generation sequencing provides information without prior knowledge of NPM1 mutation subtype or validation of allele-specific probes as required for RQ-PCR assays, and without generation and interpretation of complex multidimensional flow cytometry data. This approach may complement current technologies to enhance patient-specific clinical decision-making.
Similar content being viewed by others


Main
Acute myeloid leukemia is an aggressive hematopoietic neoplasm associated with significant mortality and morbidity. The presence of minimal residual disease, that is, small numbers of neoplastic cells which persist after cancer therapy, has been shown to correlate with disease relapse in independent cohorts, even in patients with very limited disease. Minimal residual disease detection is particularly important in individuals treated with myeloablative hematopoietic stem cell transplantation, as often these patients are younger.1, 2, 3, 4
In the United States, multiparametric flow cytometric identification of minimal residual disease is the clinical standard of care for minimal residual disease detection in acute lymphoblastic leukemia and is increasingly used at many institutions for posttreatment monitoring of acute myeloid leukemia. Flow cytometry requires detection of leukemia-associated immunophenotypes in the context of regenerative hematopoiesis, as well as complex instrumentation and analysis of high-dimensional data, making it challenging to perform routinely or on a standardized basis.5, 6 Further, as no universal immunophenotype for relapsed blasts is observed in acute myeloid leukemia, it is necessary to use a relatively large antibody panel to identify specific markers that can be used to monitor individual patients. Additionally, posttherapy immunophenotypic drift of leukemic blasts may confound consistent detection of minimal residual disease by flow cytometry, particularly in the context of marrow regeneration.7
Molecular detection of minimal residual disease by RQ-PCR has been developed recently,1, 4 and takes advantage of common mutations in the gene nucleophosmin (NPM1) in acute myeloid leukemia. Activating insertion mutations in exon 12 of NPM1 are frequent and recurrently observed in nearly one-third of all acute myeloid leukemia patients and in approximately 60% of patients with normal karyotype.8, 9 The most common mutation subtype, Type A, is a 4-bp insertion of TCTG (p.Trp288Cysfs*10, NM_002520.4:c.956_959dup). Type A mutations are frequently seen in adults, representing approximately 75–80% of NPM1 mutations in acute myeloid leukemia. However, at least 50 types of mutant NPM1 have been described,8 and some patients may have private mutant alleles. Thus, the clinical laboratory use of RQ-PCR for acute myeloid leukemia minimal residual disease diagnosis requires that the patient’s NPM1 mutation be defined in advance and that probes targeting the specific mutation be available and validated. The ability to detect unexpected NPM1 mutations may not be possible by RQ-PCR alone. Additionally, each probe differs in sensitivity and specificity, and for some mutation types probe crossreactivity with the non-mutated allele becomes limiting.4
Next-generation DNA sequencing represents an alternative molecular testing approach to detect minimal residual disease in acute myeloid leukemia. Next-generation sequencing offers significant advantages in enabling the sensitive detection of low-prevalence mutations, as well as an unprecedented economy of scale.10, 11 We recently demonstrated the first application of next-generation sequencing for the detection of minimal residual disease in T-lineage acute lymphoblastic leukemia,12 and with others showed comparable potential for minimal residual detection in B-lineage acute lymphoblastic leukemia.13, 14 Here, we describe a next-generation sequencing assay for the detection of minimal residual disease in NPM1-mutated acute myeloid leukemia.
Materials and methods
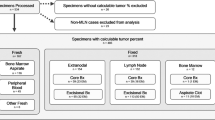
Samples
Residual, clinical samples were obtained and de-identified according to the University of Washington Institutional Review Board guidelines. A total of 22 samples from 6 patients were used in this pilot study (Table 1). All patients had a confirmed histologic or flow cytometry diagnosis of acute myeloid leukemia and were selected for further study if sufficient residual DNA (at least 200 ng) was available for analysis. DNA extraction was performed using a QiAgen X-tractor with Reagent Pack DX (Qiagen, Germantown, MD, USA).
Flow Cytometry
Multiparametric flow cytometry was performed at the University of Washington as part of the routine evaluation for minimal residual disease as described.6, 15 All samples were labeled with the antibody combinations in three tubes as follows: Tube A=HLA-DR PB, CD15 FITC, CD33 PE, CD19 ECD, CD117 PE-Cy5, CD13 PE-Cy7, CD38 A594, CD34 APC, CD71 APC-A700, and CD45 APC-H7; Tube B=HLA-DR PB, CD64 FITC, CD123 PE, CD4 ECD, CD14 PE-Cy55, CD13 PE-Cy7, CD38 A594, CD34 APC, CD16 APC-A700, CD45 APC-H7; Tube C=CD56 A488, CD7 PE, CD5 PE-Cy55, CD33 PE-Cy7, CD38 A594, CD34 APC, CD45 APC-H7. Reagents were purchased from either Becton-Dickinson (Franklin Lakes, NJ, USA) or Beckman-Coulter (Pasadena, CA, USA). Samples were processed using standard methods (red cell lysis using NH4Cl+0.25% formaldehyde), and >400 000 events were acquired on a 4-laser, 10-color Becton-Dickinson LSRII flow cytometer. Abnormal clusters of events that differed from regenerative blasts were reported relative to total leukocytes following erythroid lysis. Groups of events differing from regenerative blasts by visual inspection were identified using in-house software and considered minimal residual disease.
Next-Generation Sequencing
Sequencing library production methods were modified from Caporaso et al.16 Two hundred nanograms of DNA was amplified using KAPA HiFi HotStart ReadyMix (Kapa Biosystems, Woburn, MA, USA) with primers specific to NPM1 (reverse primer: 5′-AATGATACGGCGACCACCGAGATCTACACTATGGTGCCTGTAAACACGGTAGGGAAAGTTCTC-3′; forward primer: 5′-CAAGCAGAAGACGGCATACGAGATNNNNNNNNAGTCAGTCAGTCTGTCTATGAAGTGTTGTGGTTCC-3′, where N’s indicates the position of an 8 bp, sample-specific index sequence). Amplicons from different specimens were pooled in equimolar amounts and sequenced on an Illumina MiSeq (Illumina, San Diego, CA, USA) using 150 paired-end chemistries. Custom sequencing primers were used (Read 1: 5′-TATGGTGCCTGTAAACACGGTAGGGAAAGTTCTCA-3′; Read 2: 5′-AGTCAGTCAGTCTGTCTATGAAGTGTTGTGGTTCC-3′; Index Read: 5′-GGAACCACAACACTTCATAGACAGACTGACTGACT-3′). Oligonucleotides were synthesized by Integrated DNA Technologies (Coralville, IA, USA).
Data Analysis
Sequencing runs were demultiplexed using on-instrument software, allowing only perfectly matched index sequences to be assigned to their specimen of origin. Overlapping, paired-end reads were self-assembled using PANDAseq. 17 The length of the DNA fragment sequenced (162 bp in a non-mutated gene) enables error correction of overlapping, independently sequenced paired-end reads17 to be performed over the majority of the sequence fragment, including the documented sites of NPM1 insertion mutations. Self-assembled reads were mapped to the human genome (hg19/Ghr37) using the bwasw alignment mode of the aligner bwa (0.6.2)18 with non-default parameter ‘-r 1’ as described elsewhere.19 Variants were called using VarScan (v.2.3.6) 20 using parameter sets for a minimum variant frequency of 1 × 10−10, a minimum of 1 variant read, and a minimum average base quality of 5.
Results
Performance Characteristics of Next-Generation Sequencing and Flow Cytometry on a Defined NPM1-Positive Sample
To explore the potential of next-generation sequencing for detecting NPM1-mutant minimal residual disease, we diluted NPM1-mutated cell line OCI-AML321 into normal patient marrow (a staging specimen from a patient with classical Hodgkin lymphoma, which was negative by flow cytometry for abnormal B- and T-cell populations, myeloid blasts, and maturing myelomonocytic cells). Four separate technical replicates of each dilution were prepared for sequencing on each of three independent sequencing runs, yielding a total of 12 independent assessments per dilution. We found that semiquantitative measurement of spiked cells could be achieved over four orders of magnitude (10–0.01%; Figure 1), with minimal intra- and inter-run variability (Table 2). At a level of 0.001% NPM1-mutated cells, flow cytometry did not detect any abnormal cells. At that dilution, next-generation sequencing replicates inconsistently recovered and detected NPM1 mutation-positive reads: all four replicates from the first run were positive, whereas mutated sequence reads were detectable from only one and two replicates in the second and third sequencing runs, respectively. Thus, approximately 0.001% abnormal cells represent the limit of detection for conditions tested, comparable to the performance of RQ-PCR.4 Given an input of 200 ng DNA (corresponding to approximately 33 333 diploid cells), the assay has a theoretical detection limit of 1 cell in 33 333 (0.003%), consistent with our empirical observations when Poisson sampling from a population of low-prevalence molecules is considered. In this controlled experiment, next-generation sequencing and flow cytometry provide comparable detection of spiked OCI-AML3 cells to the level of 0.01%. Nevertheless, it should be noted that the flow cytometry diagnosis of minimal residual disease in clinical practice is typically more challenging than this contrived scenario,5, 6, 7 which used an abnormal cell line with a distinct immunophenotype (data not shown), which was simple to identify.

Next-generation sequencing for minimal residual disease detection of NPM1 mutations. Comparison of next-generation sequencing (dash) versus flow cytometry (circle) results using cell mixtures of the NPM1 mutation-positive OCI-AML3 cell line diluted into normal marrow. Graph depicts four technical replicates for each next-generation sequencing sample, with each set of technical replicates sequenced on three separate sequencing runs (red, green, or blue). Intra- and inter-run reproducibility data are shown in Table 2.
Specificity of Next-Generation Sequencing for Detection of NPM1 Mutations
We next evaluated the specificity of next-generation sequencing for detecting NPM1-mutated minimal residual disease by examining peripheral blood samples from 20 anonymous, normal blood donors. A total of 8 861 993 sequence reads were generated from that cohort, corresponding to an average of 443 100±41 438 (mean±s.d.) reads per individual. On analysis, no sequence reads matched any of the 17 most common NPM1 mutations,4 corresponding to a sensitivity of <1 in 221 550 cells (0.00045%) for detecting a false-positive mutant allele. We conclude that identification of NPM1 insertion mutations by next-generation sequencing is specific for minimal residual disease detection.
Comparison of Next-Generation Sequencing to Flow Cytometry in Longitudinal Clinical Samples
We investigated next-generation sequencing performance using 22 longitudinal clinical samples derived from six acute myeloid leukemia patients. In all samples tested, the patient’s original NPM1 mutation identified at diagnosis, which we term the ‘index clone’, could be convincingly detected. Of note, we could detect the index clone in six posttreatment samples that were interpreted as negative by clinical flow cytometry (Table 1). This finding, coupled with the high specificity of the assay, suggests that next-generation sequencing may enhance the sensitivity for minimal residual disease detection as compared with flow cytometry in clinical samples.
Detection of NPM1 Mutation Clonal Heterogeneity
In samples with detectable NPM1 mutations, it was possible to identify rare artifacts resulting from single-base substitution errors introduced through library preparation and sequencing as mutant alleles.19 In our cell line dilution studies, for which the specific NPM1 mutation is known and defined, this empiric level of error was experimentally quantified as 0.02±0.027% (mean±s.d.) of the true mutant alleles sequenced. Secondary mutations occurring significantly (several standard deviations) above that threshold, however, likely indicate the presence of other clones.
In Patient 1, an alternate clone was clearly identified. In the index sample, only one NPM1 mutation (NM_002520.6:c.(864G>C;869delinsTCCTA)) was detected. However, in four subsequent samples spanning 38 months, next-generation sequencing detected increasing levels of the Type A allele that were quantified significantly above the empiric error frequency (>3 s.d.), with a concurrent decrease of the index allele (Figures 2a and b). At days 1127 and 1162, the read counts of Type A allele surpassed those of the index allele. In Patient 2, we similarly detected heterogeneity (Figures 2a and c), harboring a Type B index mutation (NM_002520.6:c.863_864insCATG). A low prevalence, but significant level (>900 s.d. above the empiric error frequency) of Type R allele (NM_002520.6:c.863_864insTATG) was found in the index case, but became undetectable after initialization of treatment. By day 37, a low-prevalence Type A clone was identified at significant levels (>25 s.d. above the empiric error frequency), which became dominant at day 110. At ∼1 year, the amount of Type A allele was surpassed by the index clone, but nevertheless persisted.

Evidence for NPM1 mutation heterogeneity in clinical samples. (a) Multiple alignment of presentation (index) and variant subclone NPM1 mutations detected in Patients 1 and 2. Bold indicates NPM1 stop codon. (b) Detection of subclones in Patient 1 showing decline of the index clone and subsequent rise of secondary clone (Type A mutation). (c) Detection of subclones in Patient 2, showing development and low-level persistence of secondary Type A mutation. Heterogeneity is also demonstrated in the index case by the presence of a Type R allele. (d) Flow cytometry immunophenotype for Patient 1 at time-points: 0 (index), top row, day 1162, middle row, and then one additional time-point, approximately 2.5 months after day 1162, bottom row. The original index clone was an expanded monoblast population, (top row, 2nd column, asterisk) without a substantial myeloid blast population component (top row, 3rd and 4th columns). The alternate clone in subsequent samples (bottom 2 rows) no longer had a monoblastic immunophenotype but rather had an abnormal myeloid blast population with aberrant lymphoid antigen expression and accounted for 0.3% and then 1.8% of total white cells, respectively (bottom two rows, 3rd and 4th columns, arrows).
In both of these patients, our findings are most consistent with the detection of genetically distinct NPM1 clones. These alternate alleles are unlikely to be artifacts owing to the abundance of reads detected. Moreover, the Type A NPM1 mutant allele differs from the index allele seen at diagnosis for Patient 1 by 10 nucleotides and from that of Patient 2 by 2 nucleotides (Figure 2a), a degree that is unlikely to result from simultaneous sequencing artifacts in multiple reads. The interpretation of alternative clones is further supported by flow cytometry in Patient 1 (Figure 2d). Not only did the relapse myeloid blast population differ from the index monoblast clone by immunophenotype, this relapse myeloid blast population continued to increase in proportion over time (Table 1), mirroring the results for the second NPM1 clone as detected by sequencing. These results together provide evidence that next-generation sequencing can detect distinct tumor clones as defined by different NPM1 mutations. Alternate NPM1 mutations were not identified in other patients examined.
Discussion
Mutations in NPM1 have been shown to represent an early genetic lesion in acute myeloid leukemia and are therefore thought to be one of several driver mutations in this disease.8 Several groups have demonstrated that mutant NPM1 is generally stable through treatment, and typically, NPM1 mutation can be detected in posttreatment relapse samples as a measure of recurrent disease.22 Some have suggested that mutant NPM1 is not identified in relapse samples from a small subset of patients, implying that mutation in NPM1 is not always an early lesion or that NPM1-mutated cells may respond to treatment.23, 24 Nevertheless, the prognostic value of monitoring NPM1 mutations in minimal residual disease has been independently demonstrated.4, 25 Of particular note, NPM1 mutation level has been confirmed as an independent clinical prognostic factor for relapse and survival.26 Thus, monitoring of acute myeloid leukemia through NPM1 mutation is generally agreed to be an important prognostic marker.
The most widely used approaches for detecting minimal residual disease in NPM1-mutated acute myeloid leukemia, in general, are RQ-PCR1, 4 and flow cytometry.2, 3 However, as each approach offers its own combination of benefits and drawbacks, the best method to assess minimal residual disease in a patient care context remains a matter of debate.27, 28, 29 Importantly, it should be noted that in limited studies in which molecular diagnosis of various mutated targets and flow cytometry were directly compared, results have demonstrated equivalency of these approaches,27 and it is generally believed that both RQ-PCR and flow cytometry are suitable for diagnosis of minimal residual disease in acute myeloid leukemia.28, 30 In this work, we explored the utility of next-generation sequencing to identify minimal residual disease in NPM1-mutated acute myeloid leukemia and report several key findings.
The first major finding of this work is that although next-generation sequencing has proven only semiquantitative, likely secondary to PCR bias during sequencing library construction, the use of next-generation sequencing overcomes several of the limitations presented by both flow cytometry and RT-PCR for minimal residual disease detection. As compared with flow cytometry, we found that one advantage of minimal residual disease detection by next-generation sequencing is the potential for increased sensitivity for detecting abnormal blasts. In our cell dilution studies, next-generation sequencing was determined to have a limit of detection of approximately 0.001% minimal residual disease, an order of magnitude greater than observed in concurrent flow cytometry studies for the conditions tested (Figure 1). Even so, consistent clinical detection of abnormal myeloid blasts and particularly blast equivalents (promonocytes or monoblasts) by flow cytometry is often more challenging than this contrived experimental scenario, and the level of success depends greatly upon on the immunophenotype of the abnormal blasts and how distinct it is from background regenerative blasts.5, 6, 7 This was also observed in our analysis of clinical samples. Specifically, it is noteworthy that all clinical specimens examined in this study, even if diagnostically negative by flow cytometry, were found to harbor minimal residual disease by next-generation sequencing (Table 1). The clinical implication of these low-frequency positive results though merits further study and consideration. Some investigators have postulated that the persistence of low levels of minimal residual disease in some patients may indicate effective immune surveillance,27 whereas others have suggested that these results may alternatively represent an early harbinger of relapse.31 Irrespective, the consistent detection of minimal residual disease by next-generation sequencing after treatment in 6 of 6 randomly selected patients of our cohort suggests that low-level clonal persistence may be a consistent biological feature of acute myeloid leukemia. Accordingly, monitoring of patients by next-generation sequencing may provide important clinical information regarding the kinetics of these populations and thus insight into the biologic and clinical behavior of a patient’s disease.
Another advantage of next-generation sequencing as compared to flow cytometry is the potential for enhanced specificity. While in general the level of blasts estimated by next-generation sequencing was comparable to that of flow cytometry, in a subset of cases, the measured leukemic blast proportion by next-generation sequencing differed from flow cytometry-based estimates (Table 1). There are two important points to note regarding these apparent discordances. First, by flow cytometry, admixed regenerative myeloid blasts may be variably present in the posttreatment setting and consequently difficult to distinguish consistently from leukemic blasts owing to an overlap of immunophenotypes between the two populations. This may therefore lead to either over- or underestimation of leukemic blasts by flow cytometry, depending on how gating of the leukemic blast versus regenerative blast populations is performed. Second, as flow cytometry is typically performed on samples subjected to erythroid lysis protocols that also result in the removal of nucleated erythroid precursors, the blast estimate by flow cytometry is enumerated and reported with respect to total white cells. However, DNA from early erythroid precursors will contribute to the total templates examined by DNA sequencing. Thus, in cases with erythroid hyperplasia, flow cytometry estimates of blast percentage may be greater than those estimated by sequencing. As next-generation sequencing is not subject to either of these artifacts, it is likely that next-generation sequencing more accurately estimates the minimal residual disease burden, albeit in a semiquantitative manner. This conclusion is supported by our cell spike-in studies (Figure 1), in which we observed strong concordance of next-generation sequencing results versus theoretical dilutions for NPM1 mutation detection.
As compared with RT-PCR, there are several potential advantages to the next-generation sequencing approach we propose herein. As the primers used in our assay are situated outside of NPM1 exon 12, all reported NPM1 mutations9 should be detectable with comparable efficiency using this approach. Accordingly, this assay eliminates the need for mutation-specific probes as required for RQ-PCR and the related challenges in ensuring suitable performance characteristics for multiple probe sets.4, 30 As a case in point, Patient 1 had a novel mutation (NM_199185.3:c.777_782delinsCCAGTTCCTA), previously unreported in COSMIC (Figure 2a). This contrasts with RQ-PCR assays that require prior knowledge of the NPM1 mutation subtype or use of multiple mutation-specific probes.4 In light of these considerations, next-generation sequencing for minimal residual disease could serve as a universal approach for clinical laboratory detection in NPM1-mutated acute myeloid leukemia, providing a technically defined analytic method that is equally applicable to all patients with NPM1-mutated blasts. Notably, both RQ-PCR and our proof-of-principle next-generation sequencing assay are still limited to use in cases in which NPM1 mutation-positive minimal residual disease is present. However, because next-generation sequencing technologies permit high-throughput analysis with enhanced economy of scale, next-generation sequencing-based assays could be readily expanded to include additional potentially informative targets, such as DNMT3A, KIT, IDH1, IDH2 or other driver mutations, for minimal residual disease detection.32, 33, 34, 35, 36, 37
As we have previously noted, the sensitivity of our assay was similar to published values for RQ-PCR assays.4 Nevertheless, minor modifications could increase this value even further. The sensitivity with which next-generation DNA sequencing is able to detect low-prevalence mutations is proportional to the number of DNA molecules examined, the effective number of sequence reads generated for each template molecule, and the rate with which sequencing or library preparation errors result in false-positive mutation calls.19, 38 Here, the specificity of the assay for NPM1 insertion mutations has proven non-limiting, with no false-positive NPM1 mutation reads detected in normal blood donor samples: this specificity is due to the fact that it is quite unlikely by PCR or sequencing artifact to replicate 4 bp or larger NPM1 insertion mutations. Accordingly, it should theoretically be possible to further increase sensitivity by proportionally scaling the amount of template DNA and the read depth allocated per sample, assuming that sufficient quantities of sample are available. We also note that, while others previously explored the use of next-generation sequencing for detecting NPM1 mutations, the reported limit of detection for those studies was 0.02%,11 20-fold higher than reported here. Aside from technical considerations including differences in sequencing platforms used, this difference likely resulted in part from undersampling of template alleles, given the limited read depth (∼7758 reads). Thus, some reported negative results in the previous study may represent false negatives.11
The second major finding of this study is the presence of alternate, genetically distinct NPM1-positive subclones that were identified in a surprisingly high proportion (2 of 6) of the randomly selected acute myeloid leukemia patients we studied. In Patient 1 (Figure 2b), the original, index clone was seen to decrease after initiation of therapy, but a secondary population bearing a different NPM1 mutation gradually increased in proportion, coinciding with the detection of an immunophenotypically distinct population of abnormal blast by flow cytometry (Figure 2d). In the other, Patient 2 (Figure 2c), an alternate subclone was detected at the time of diagnosis, but was not identified in other specimens from the same patient, suggesting its elimination during treatment. Nevertheless, a separate, genetically distinct clone was detected just over 1 month after the index sample and persisted through the remaining two time points. Intriguingly, it was the patient’s original index clone, and not either subclone, that returned during the patient’s apparent relapse a year after diagnosis. Findings from this unselected, albeit limited cohort suggest both that the dynamics of minimal residual disease and acute myeloid leukemia relapse may be more complex than previously appreciated and that subclones may occur in an apparently significant proportion of acute myeloid leukemia patients. The identification of such subclones has not previously been possible by existing clinical technologies, although recent work has suggested that acute myeloid leukemia clonal heterogeneity can represent an adverse prognostic marker in some patients.39 Longitudinal studies will be required to determine the clinical significance of clonal heterogeneity detected by next-generation sequencing in NPM1-mutated acute myeloid leukemia.
Lastly, the clinical detection of uncommon (non-Type A) alleles of NPM1 may be clinically important and is efficiently achieved by next-generation sequencing. Alpermann et al40 recently reported that rare NPM1 mutation types had a trend toward favorable event-free and overall survival as compared with patients with the common Type-A mutations. Moreover, in patients with concurrent FLT3-ITD mutation, which is typically regarded as a poor prognostic genetic marker that over-rides the favorable prognostic effect of NPM1 mutation, only patients with Type A NPM1 mutations showed poor overall survival, with uncommon NPM1 mutations conferring significantly better event-free and overall survival.40 Next-generation sequencing offers a robust approach to ascertain the specific NPM1 mutation present and the presence of subclones or relapse clones bearing prognostically different NPM1 mutations simultaneously.
In summary, the favorable performance characteristics and expanded diagnostic capabilities of next-generation sequencing suggest that this method may enhance minimal residual disease detection in NPM1-mutated acute myeloid leukemia, augmenting technologies for minimal residual disease detection currently in clinical use. In this regard, next-generation sequencing may permit opportunities for earlier clinical intervention, either through detecting very low levels of minimal residual disease, the detection of acute myeloid leukemia heterogeneity, or by enabling monitoring of the rate of change in minimal residual disease abundance.31 Defining the clinical significance of low-level minimal residual disease by next-generation sequencing, however, remains an important task.
References
Bacher U, Badbaran A, Fehse B et al. Quantitative monitoring of NPM1 mutations provides a valid minimal residual disease parameter following allogeneic stem cell transplantation. Exp Hematol 2009;37:135–1342.
Walter RB, Buckley SA, Pagel JM et al. Significance of minimal residual disease before myeloablative allogeneic hematopoietic cell transplantation for AML in first and second complete remission. Blood 2013;122:1813–1821.
Walter RB, Gooley TA, Wood BL et al. Impact of pretransplantation minimal residual disease, as detected by multiparametric flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute myeloid leukemia. J Clin Oncol 2011;29:1190–1197.
Schnittger S, Kern W, Tschulik C et al. Minimal residual disease levels assessed by NPM1 mutation-specific RQ-PCR provide important prognostic information in AML. Blood 2009;114:2220–2231.
Kern W, Voskova D, Schoch C et al. Determination of relapse risk based on assessment of minimal residual disease during complete remission by multiparameter flow cytometry in unselected patients with acute myeloid leukemia. Blood 2004;104:3078–3085.
Wood BL. . Flow cytometric monitoring of residual disease in acute leukemia. Methods Mol Biol 2013;999:123–136.
Voskova D, Schoch C, Schnittger S et al. Stability of leukemia-associated aberrant immunophenotypes in patients with acute myeloid leukemia between diagnosis and relapse: comparison with cytomorphologic, cytogenetic, and molecular genetic findings. Cytometry B 2004;62:25–38.
Falini B, Martelli MP, Bolli N et al. Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood 2011;117:1109–1120.
Falini B, Mecucci C, Tiacci E et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 2005;352:254–266.
Pritchard CC, Salipante SJ, Koehler K et al. Validation and implementation of targeted capture and sequencing for the detection of actionable mutation, copy number variation, and gene rearrangement in clinical cancer specimens. J Mol Diagn 2014;16:56–67.
Thol F, Kolking B, Damm F et al. Next-generation sequencing for minimal residual disease monitoring in acute myeloid leukemia patients with FLT3-ITD or NPM1 mutations. Genes Chromosomes Cancer 2012;51:689–695.
Wu D, Sherwood A, Fromm JR et al. High-throughput sequencing detects minimal residual disease in acute T lymphoblastic leukemia. Sci Transl Med 2012;4:134ra63.
Faham M, Zheng J, Moorhead M et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood 2012;120:5173–5180.
Wu D, Emerson RO, Sherwood A et al. Detection of minimal residual disease in patients with B lymphoblastic leukemia by high-throughput sequencing of IGH. Submitted.
Wood B . 9-color and 10-color flow cytometry in the clinical laboratory. Arch Pathol Lab Med 2006;130:680–690.
Caporaso JG, Lauber CL, Walters WA et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 2012;6:1621–1624.
Masella AP, Bartram AK, Truszkowski JM, Brown DG, Neufeld JD . PANDAseq: paired-end assembler for illumina sequences. BMC Bioinform 2012;13:31.
Li H, Durbin R . Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010;26:589–595.
Hiatt JB, Pritchard CC, Salipante SJ, O'Roak BJ, Shendure J . Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res 2013;23:843–854.
Koboldt DC, Zhang Q, Larson DE et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 2012;22:568–576.
Quentmeier H, Martelli MP, Dirks WG et al. Cell line OCI/AML3 bears exon-12 NPM gene mutation-A and cytoplasmic expression of nucleophosmin. Leukemia 2005;19:1760–1767.
Kronke J, Schlenk RF, Jensen KO et al. Monitoring of minimal residual disease in NPM1-mutated acute myeloid leukemia: a study from the German–Austrian acute myeloid leukemia study group. J Clin Oncol 2011;29:2709–2716.
Suzuki T, Kiyoi H, Ozeki K et al. Clinical characteristics and prognostic implications of NPM1 mutations in acute myeloid leukemia. Blood 2005;106:2854–2861.
Kronke J, Bullinger L, Teleanu V et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood 2013;122:100–108.
Hafez M, Ye F, Jackson K et al. Performance and clinical evaluation of a sensitive multiplex assay for the rapid detection of common NPM1 mutations. J Mol Diagn 2010;12:629–635.
Shayegi N, Kramer M, Bornhauser M et al. The level of residual disease based on mutant NPM1 is an independent prognostic factor for relapse and survival in AML. Blood 2013;122:83–92.
Paietta E . Minimal residual disease in acute myeloid leukemia: coming of age. Hematology Am Soc Hematol Educ Program 2012;2012:35–42.
Buccisano F, Maurillo L, Del Principe MI et al. Prognostic and therapeutic implications of minimal residual disease detection in acute myeloid leukemia. Blood 2011;119:332–341.
Shook D, Coustan-Smith E, Ribeiro RC, Rubnitz JE, Campana D . Minimal residual disease quantitation in acute myeloid leukemia. Clin Lymphoma Myeloma 2009;9 (Suppl 3):S281–S285.
Grimwade D, Vyas P, Freeman S . Assessment of minimal residual disease in acute myeloid leukemia. Curr Opin Oncol 2010;22:656–663.
Freeman SD, Jovanovic JV, Grimwade D . Development of minimal residual disease-directed therapy in acute myeloid leukemia. Semin Oncol 2008;35:388–400.
Ley TJ, Minx PJ, Walter MJ et al. A pilot study of high-throughput, sequence-based mutational profiling of primary human acute myeloid leukemia cell genomes. Proc Natl Acad Sci USA 2003;100:14275–14280.
Conte N, Varela I, Grove C et al. Detailed molecular characterisation of acute myeloid leukaemia with a normal karyotype using targeted DNA capture. Leukemia 2013;27:1820–1825.
Mardis ER, Ding L, Dooling DJ et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 2009;361:1058–1066.
Welch JS, Ley TJ, Link DC et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012;150:264–278.
Ding L, Ley TJ, Larson DE et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012;481:506–510.
Network TCGAR. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–2074.
Harismendy O, Schwab RB, Bao L et al. Detection of low prevalence somatic mutations in solid tumors with ultra-deep targeted sequencing. Genome Biol 2011;12:R124.
Bochtler T, Stolzel F, Heilig CE et al. Clonal heterogeneity as detected by metaphase karyotyping is an indicator of poor prognosis in acute myeloid leukemia. J Clin Oncol 2013;31:3898–3905.
Alpermann T, Haferlach C, Dicker F et al. Evaluation of different NPM1 mutations in AML patients according to clinical, cytogenetic and molecular features and impact on outcome. Blood 2013;122:51.
Acknowledgements
We thank A Thomas, UW Hematopathology staff, C Pritchard and J Tait, for technical help and support of this work. JS is funded by Grant CA160080 from the National Cancer Institute. This work was supported by the Department of Laboratory Medicine, University of Washington.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Salipante, S., Fromm, J., Shendure, J. et al. Detection of minimal residual disease in NPM1-mutated acute myeloid leukemia by next-generation sequencing. Mod Pathol 27, 1438–1446 (2014). https://doi.org/10.1038/modpathol.2014.57
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2014.57
Keywords
This article is cited by
-
Clinical impact of panel-based error-corrected next generation sequencing versus flow cytometry to detect measurable residual disease (MRD) in acute myeloid leukemia (AML)
Leukemia (2021)
-
NPM1 Biology in Myeloid Neoplasia
Current Hematologic Malignancy Reports (2020)
-
Clinical Validation and Implementation of a Measurable Residual Disease Assay for NPM1 in Acute Myeloid Leukemia by Error-Corrected Next-Generation Sequencing
Molecular Diagnosis & Therapy (2019)
-
Methods of Detection of Measurable Residual Disease in AML
Current Hematologic Malignancy Reports (2017)
-
Quantifying ultra-rare pre-leukemic clones via targeted error-corrected sequencing
Leukemia (2015)
