
Abstract
Massively parallel sequencing (MPS) has become a viable diagnostic tool to interrogate genetic profiles of numerous tumors but has yet to be routinely adopted in the setting of lymphoma. Here, we report the empirical application of a targeted 40-gene panel developed for use in mature lymphoid neoplasms (MLNs) and report our experience on over 500 cases submitted for MPS during the first year of its clinical use. MPS was applied to both fresh and fixed specimens. The most frequent diagnoses were diffuse large B-cell lymphoma (116), chronic lymphocytic leukemia/small lymphocytic lymphoma (60), marginal zone lymphoma (52), and follicular lymphoma (43), followed by a spectrum of mature T-cell neoplasms (40). Of 534 cases submitted, 471 generated reportable results in MLNs, with disease-associated variants (DAVs) detected in 241 cases (51.2%). The most frequent DAVs affected TP53 (30%), CREBBP (14%), MYD88 (14%), TNFRSF14 (10%), TNFAIP3 (10%), B2M (7%), and NOTCH2 (7%). The bulk of our findings confirm what is reported in the scientific literature. While a substantial majority of mutations did not directly impact diagnosis, MPS results were utilized to either change, refine, or facilitate the final diagnosis in ~10.8% of cases with DAVs and 5.5% of cases overall. In addition, we identified preanalytic variables that significantly affect assay performance highlighting items for specimen triage. We demonstrate the technical viability and utility of the judicious use of a targeted MPS panel that may help to establish general guidelines for specimen selection and diagnostic application in MLNs in routine clinical practice.
Similar content being viewed by others

Introduction
Mature lymphoid neoplasms (MLNs) are a leading cause of cancer mortality worldwide and represent an extensive, clinically diverse, and pathologically heterogeneous group of entities. These neoplasms have been classified based upon numerous factors including cell type (B-cell vs. T-cell), degree of maturity (precursor vs. mature), anatomic site (nodal, splenic, cutaneous, etc.), predicted natural history (indolent vs. aggressive), and underlying disease (immunodeficiency, etc.), amongst others. These granular degrees of categorization are incorporated into the updated 2017 WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues [1]. The finer degrees of separation for these entities provide not only a better understanding of the distinct mechanistic biology of each disease process, but also allow for improved clinical insight into management strategies and prognosis. Genetic testing has played an important role in the evaluation of MLNs, primarily at the level of chromosomal aberrations, with the advent of sophisticated genomic approaches further unraveling unprecedented insights into the pathobiology of these neoplasms.
Although mutational profiling of hematologic neoplasms currently predominates in the area of myeloid neoplasms, there is a growing wealth of literature describing common and highly characteristic genetic alterations within the individual subcategories of MLNs [2,3,4,5,6,7]. For example, the diagnostic approach to entities such as lymphoplasmacytic lymphoma (LPL) and hairy cell leukemia (HCL) has been altered with the discovery of MYD88 mutations in ~90% of LPL cases and BRAF V600E mutations in nearly all HCL cases [8, 9]. Beyond the consideration of these characteristic mutations, enriched genetic profiles with diagnostic, prognostic, and therapeutic relevance have been observed in the vast array of these neoplasms [10]. Common disease-associated mutations have been identified in small mature B-cell neoplasms including: chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) [e.g., SF3B1, TP53, NOTCH1, ATM], follicular lymphoma (FL) [e.g., CREBBP, TNFRSF14, TNFAIP3, EZH2], mantle cell lymphoma (MCL) [e.g., ATM, CCND1, TP53, NOTCH2, BIRC3], and marginal zone lymphomas (MZL) [e.g., KLF2, NOTCH2, TNFAIP3, TRAF3] [11]. Additional studies have described the most frequent mutations in aggressive B-cell neoplasms, including germinal center B-cell (GCB) diffuse large B-cell lymphoma (DLBCL) [e.g., EZH2, GNA13, TNFRSF14, SOCS1], activated B-cell (ABC) DLBCL [e.g., MYD88, CD79A/B, CARD11, TNFAIP3], and Burkitt lymphoma [e.g., TCF3, ID3] [12]. Furthermore, individual subcategories of lymphoid neoplasms tend to display recurrent mutational profiles. These include those affecting MYD88 in primary DLBCL of the CNS, TNFAIP3, STAT6, XPO1, and SOCS1 in primary mediastinal large B-cell lymphoma (PMBL), TET2, DNMT3A, RHOA, and IDH2 in angioimmunoblastic T-cell lymphoma (AITL), and STAT3 in T-cell large granular lymphocyte leukemia [13,14,15,16]. As noted, certain mutations are enriched in specific categories of MLNs, and new discoveries continue to emerge at a rapid pace. For example, comprehensive restructuring of the categorization of DLBCL subtypes from beyond the cell of origin theory and numerical “hit” hypotheses to a clustering paradigm based on genetic signatures that correspond with prognostic and therapeutic significance has been proposed [17, 18]. In addition, alternative perspectives have been suggested for the utilization of genetic profiles as biomarkers for prognosis, diagnostics, targeted therapeutics, and response to therapy across many of the other categories of MLNs [19,20,21].
The rapid evolution of massively parallel sequencing (MPS), also referred to as next generation or high throughput sequencing, has led to the development of strategies for clinical utilization of comprehensive tumor profiling information which enables personalized medicine [22]. Qualitative and quantitative “single gene” assays remain relevant to identify certain pathognomonic, prognostically useful, and therapeutically implicated mutations in hematologic neoplasms. In myeloid disease, single gene testing for, amongst others, JAK2, NPM1, CEBPA, FLT3, IDH1/2, and KIT mutations, and BCR-ABL1 fusions are useful for diagnosis and therapy, while single gene assays for MYD88 L265P and BRAF V600E mutations are widely utilized for the assessment of LPL and HCL, respectively [23]. However, targeted MPS panels provide an opportunity to interrogate numerous genes simultaneously, serving to restructure diagnostic and therapeutic paradigms [24,25,26].
Despite the impressive large-scale genomic characterization of MLNs uncovered by numerous sophisticated scientific discoveries, exploration of the clinical and practical diagnostic utility of a targeted MPS panel for use in MLNs has only very recently undergone limited scrutiny [27]. Our institution applied a targeted 40-gene MPS panel for MLNs capable of detecting single-nucleotide variants (SNVs) and small insertion/deletions (indels) to a series of specimens over a 1-year period. We report on our institutional experience, the largest to date, specifically focusing on the diagnostic utility and technical performance of this panel, with the aim of validating its clinical utility and identifying preanalytic variables that influence outcomes, with a view to providing some guidance to the use of such panels in the routine clinical realm.
Materials and methods
Study design and patient characteristics
We reviewed a total of 598 sequential MPS lymphoma panel orders between November 1, 2018 and October 31, 2019, representing samples from 518 unique patients. The orders were placed entirely at the discretion of the treating clinician, namely the hematologist–oncologist caring for the patient, or the hematopathologist responsible for rendering a definitive diagnosis. A total of 41 patients had more than one study performed, which accounted for 85 (14.2%) of the MPS panel orders. Common clinical reasoning for multiple studies on a single patient included bone marrow staging at diagnosis of an extramedullary MLN, subsequent bone marrow evaluation of residual disease after treatment and/or bone marrow transplant, and repeat testing performed on samples with initial quantitative or qualitative sequencing failure. During this time period, ~942 unique patients with newly diagnosed or relapsed MLN were seen by clinicians in the Lymphoma Program in the Division of Hematology-Oncology in the Department of Medicine at the Hospital of the University of Pennsylvania. Patient and pathologic information was collected from the electronic medical record (EMR) and laboratory information system (LIS) under an approved retrospective IRB protocol.
Massively parallel sequencing
Detection of SNVs and insertions/deletions of less than 25 base pairs was performed by the University of Pennsylvania Center for Personalized Diagnostics in the Department of Pathology and Laboratory Medicine and Abramson Cancer Center. Acceptable specimens for evaluation included blood, bone marrow, fresh tissue, fine needle aspirates (FNAs), and formalin-fixed paraffin-embedded (FFPE) tissue, with a minimum requirement of 10% tumor nuclei within the specimen. DNA was extracted from fresh tissue, FNA specimens, and FFPE tissue using an Agencourt FormaPure Kit (Beckman Coulter; Brea, CA) and from blood and bone marrow specimens using the DSP DNA Mini kit (Qiagen; Germantown, MD). DNA was quantified with a Qubit dsDNA BR Assay (Invitrogen). Individual libraries were prepared using a TruSight Lymphoma 40 kit (Illumina; San Diego, CA) and TapeStation analysis (Agilent) was performed to demonstrate successful library preparation prior to sequencing. Enriched library preparations, in duplicate per patient, were pooled and sequenced in parallel on a MiSeq (Illumina) platform using multiplexed, paired-end reads, each with a mean read depth of ~5500–6000 per pool. As a quality control metric, 250 reads passing filter per pool at a given locus was required to call a variant. Analysis and interpretation were performed using the Illumina Amplicon DS module (lymphoma_v1) with reference to the hg19 Genome build. Categorization of variants was dependent upon review of the literature, the presence of the variant in publicly available databases including dbSNP, COSMIC, gnomAD, the 1000 genome project, cBioPortal, and VarSome. The genes sequenced were part of a commercially available targeted MPS amplicon panel for 40 clinically and diagnostically relevant lymphoma-associated gene variants that included: ATM, B2M, BIRC3, BRAF, BTK, CARD11, CD79A, CD79B, CIITA, CREBBP, CXCR4, EGR2, EZH2, GNA13, ID3, IDH2, JAK3, KLF2, MAP2K1, MYD88, NFKBIE, NOTCH1, NOTCH2, PLCG1, PLCG2, POT1, RHOA, RPS15, RRAGC, SF3B1, SOCS1, STAT3, STAT5B, TCF3, TET2, TNFAIP3, TNFRSF14, TP53, TRAF3, and XPO1. The full coding region for ATM, CREBBP, CXCR4, GNA13, JAK3, MYD88, SOCS1, TNFRSF14, and TP53 were covered by the assay. The remaining genes were partially covered with minimal coverage of known clinically relevant hotspots known at the time of the assay design. Analysis was determined to be a MPS failure due to either insufficient DNA, defined as quantitative failure (QTF), or inadequate DNA, defined as qualitative failure (QLF).
Tumor percentage estimation
Tumor percentage for fixed samples was estimated by the diagnosing pathologist based on morphology of H&E stained sections and if applicable, immunohistochemical stains. The reported tumor percentages were obtained by reviewing sequencing requisition forms and pathology reports. Tumor percentages were reported as discrete values and as ranges. To accommodate these differences, tumor percentages were categorized as follows ≥50%, 49–26%, 25–10%, 1–9%, and 0%. In most fixed tissue cases, reported tumor percentages fell into these predefined categories. The tumor percentage for FNAs was determined by morphology and/or flow cytometry. Few bone marrow aspirates or peripheral blood specimens had reliable tumor percentage reported. Thus, for the majority of these specimen types, tumor percentage was calculated by multiplying the percent lymphocytes on 200-cell differential by the percent neoplastic lymphocytes identified on flow cytometric evaluation. Samples for which an estimate of tumor percentage was precluded by lack of tandem flow cytometry (defined as within 30 days of sequencing), or by nonrepresentative flow cytometry, were excluded from the logistic regression analyses.
Data analysis
All patient data including basic identifying information, pertinent specimen descriptors (including whether the specimen actually contained an MLN, and if so, the percentage of tumor, whether the specimen was fixed or fresh, the nature of the biopsy such as whether it was a needle core biopsy or excisional biopsy, and whether the material was collected and processed intramurally or extramurally), hematopathologic diagnoses, pathology report addenda, and MPS results were extracted from the EMR and LIS and collated using Excel. Descriptive statistics were used to characterize these data (mean, percentage, range, frequency). Figures and tables were generated using basic charting tools in Excel. The OncoPrint was produced using the OncoPrinter tool available through the cBioPortal for Cancer Genomics website [28, 29].
Logistic regression analysis
In order to determine how preanalytic variables impact MPS results, we collected information on processing institution (intramural or submitted extramural), specimen anatomic source, specimen type (for example nature of biopsy), tumor percentage, and fixation for each specimen (Fig. 1). We constructed two separate logistic regression models using GraphPad Prism to estimate odds ratios, 95% confidence intervals, and two-tailed p values. The first analysis was modeled to determine the effect of preanalytic variables on MPS failure (QTF and QLF) as compared with success defined as a MPS result of disease-associated variant (DAV), variant of uncertain significance (VUS), or no variant (NV). The second analysis was modeled to determine potential “false negative” MPS results (defined as pathologic diagnosis present with NV MPS results). Potential “false negatives” are denoted with quotation marks to acknowledge that the MPS panel is not absolutely comprehensive for all possible mutations in MLNs.

Preanalytic variables among processed MLN specimens (n = 534) that include specimens inadequate for sequencing due to QTF or QLF, excluding non-MLN cases (n = 23) and cases in which tumor percent was not reported or not calculable (n = 26) yielding 485 MLN cases with calculable tumor percentages. Noted in parentheses is the number of extramural (EM) cases. There were no extramural fresh specimens. To facilitate statistical analysis, specimen types were grouped based on overall expected tissue quantity. “Core Bx” includes mostly core biopsies (including a single decalcified bone marrow core biopsy [see Table 1 footnote f for more detail]), seven punch biopsies, and a single FNA cell block. “Excisional Bx” includes mostly excisional biopsies of lymph nodes as well as larger excisions of extranodal tissue and a single wedge biopsy of lung tissue. “Other Fresh” includes lymph node FNA (n = 2), extranodal FNA (n = 1), and other body fluid specimens (n = 3).
Categorical preanalytic variables were recoded into binomial factors and tested in multivariate logistic regression. For recoding, the reference variable for the first analysis was selected as the variable with the lowest rate of QTF or QLF MPS and for the second analysis the variable with the lowest rate of NV MPS results was selected as the reference variable. As MPS failure was not observed in any fresh specimens tested, only fixed samples were included in the first analysis interrogating QTF and QLF MPS. Amongst fixed specimens, tumor percentages were uniformly >10%. In addition, we combined tumor percent categories of 10–25% and 25–49% as only 3 of 40 MPS failures were observed in the 10–25% tumor category.
In the second logistic regression analysis evaluating potential “false negative” MPS results, we limited the analysis to samples with pathologic diagnosis present and samples that did not fail MPS (i.e., MPS result of DAV, VUS, and NV). Fresh FNA specimens were also excluded because there were no samples in this category with a NV MPS result. Due to overlap between specimen type and source in the fresh samples, preanalytic variables of fixed/fresh, specimen type, and specimen source were combined in this analysis to prevent linearity and perfect separation.
Diagnostic utility determination
The final diagnosis rendered for each case (after comprehensive incorporation of cytogenetic, fluorescence in-situ hybridization/FISH, antigen receptor gene rearrangement/ARGR, and MPS studies, along with clinical correlation) was compared to the initial diagnosis (rendered by morphologic and immunophenotypic evaluation alone), with specific focus on the impact of MPS results, when MPS was determined to provide greater diagnostic value over and above that provided by other genetic studies. The pathology reports of all MLNs with reportable MPS results were reviewed, with the primary data for this analysis being addenda generated to original reports with comments discussing the relevance of MPS results to final diagnostic considerations. It should be noted that there was no predetermined standard of practice set for reporting the results of MPS in addenda prior to collection of these data. Hence, in cases in which addenda were not generated, the impact of MPS results on the diagnosis was reviewed and documented by two of us [ARD, AB]. The effects of MPS results on final diagnoses were classified into three categories: (1) change a diagnosis, (2) refine a differential diagnosis, and (3) facilitate a diagnosis. A change of diagnosis (1) was considered only if the initial diagnosis was reported confidently, with subsequent change to an entirely different diagnosis based on the results of MPS. MPS results were considered to refine a diagnosis (2) if the initial case had a differential listed, including reactive vs. neoplastic, with the results of MPS favoring one of the considerations listed in the differential diagnosis. The results of MPS were classified to facilitate the diagnosis (3) if the initial case had a favored but equivocal diagnosis, with the MPS results allowing for a more confident diagnosis.
Results
Specimen and result category data
The orders for 64 of the 598 cases (10.7%) were canceled prior to processing for MPS. Identifiable sources for cancellation included order errors, duplicate orders, and/or lack of necessity based on variable clinical or hematopathology triage and review. Of the remaining 534 cases that were processed (Fig. 2), 40 were not sequenced as the extracted DNA was either quantitatively (QTF) or qualitatively (QLF) suboptimal. Of the remaining 494 specimens, 23 that did not represent MLN specimens were excluded. Amongst the remaining 471 MLN specimens analyzed, DAVs were detected in 51.2%, VUSs in 17.8%. and NV in 31%. Notably, the 31% of these 471 cases with NV results includes 97 cases (20.6% of total) which yielded no mutations despite harboring a morphologically detectable MLN, and 49 cases (10.4% of total) which contained no microscopic evidence of disease and unsurprisingly yielded no mutations. For the stratification of specimens by preanalytic variables (Fig. 1), 26 cases (24 peripheral blood specimens, 2 bone marrow aspirates) were excluded because tumor percentage was not calculable. The 485 specimens with calculable tumor percentage in Fig. 1 reflect the 471 cases from Fig. 2 that were analyzed for diagnostic impact of MPS results, with the 40 cases “inadequate for sequencing” added back and the 26 cases “without calculable tumor percentage” removed.

All orders for the MPS lymphoma panel were collected for the defined study period. Canceled orders (64) were removed prior to case analysis, yielding 534 cases that were processed. Of the cases processed, 40 were either quantitatively or qualitatively insufficient for sequencing and of the sequenced cases, 23 were not representative of mature lymphoid neoplasms. Ultimately, 471 sequenced cases were analyzed of which 51.2% had detectable DAVs, 17.8% had VUSs only detected, and 31% had no variants (NV) detected. The latter cases with no variants detected were further stratified by the presence or absence of a mature lymphoid neoplasm in the specimen. Approximately one-third of the cases (49/146) with no variants (NV) detected had no disease present.
Preanalytic variables and assay performance
The first logistic regression analysis (Table 1) was designed to evaluate the impact of preanalytic variables on MPS failure or success as previously defined. We limited our analysis to fixed cases because MPS failure was not observed in fresh specimens (n = 167). While specimen type and source had little impact on MPS failure, specimens with lower tumor percent and extramural specimens were significantly more likely to result in MPS failure based on calculated odds ratios. For example, extramural specimens were 5.25 times more likely to fail MPS. Similarly, specimens with <50% tumor were 2.22 times more likely to have QTF or QLF MPS results.
Diagnostic categories
WHO 2017 criteria were used for the final diagnostic categories in all cases by incorporating initial diagnostic considerations (based primarily on morphology and immunophenotypic information) with comprehensive review of subsequent cytogenetic, FISH, ARGR, and MPS studies, as well as clinical correlation. The final diagnostic groups for all cases with corresponding MPS result categories are summarized in Table 2. The role of MPS results in affecting final diagnostic categorization is detailed in the “Diagnostic utility of MPS” section below. Of note, 61 cases (13% of total 471 analyzed) had no overt microscopic (and, where tested, no immunophenotypic) evidence of an MLN—of which 49 cases had no mutations detected, 3 bone marrow specimens were found to have DAVs of SF3B1 only, and 9 cases revealed VUSs. The rationale for submission of these morphologically and immunophenotypically negative specimens incorporated three categories: evaluation of potential residual disease (n = 9), staging of bone marrow involvement by an extramedullary process (n = 14), and clinical suspicion for involvement of the submitted specimen by an MLN (n = 38).
MPS results
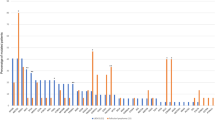
Overall, 35 of the 40 genes on the panel were identified at least once as a DAV, while 3 of the genes presented with a VUS only (CARD11, RRAGC, and TCF3), and no genetic alterations were observed in PLCG1 or PLCG2. The most frequent disease-associated genetic alterations across all diagnostic categories were identified in TP53, MYD88, CREBBP, and EZH2 at 30%, 14%, 14%, and 12%, respectively, (Fig. 3). DAVs in the remaining genes on the panel occurred at a frequency of 10% or less: NOTCH1, TNFAIP3, and TNFRSF14 (10% each), and B2M and NOTCH2 (7% each). Four genes were mutated at a frequency of 5% (ATM, BRAF, NFKBIE, and SOCS1). The remaining genes on the panel with DAVs had a mutation frequency of less than 5%. Table 3 details the frequency of mutations present for each gene stratified by diagnostic subcategory. The total number of DAVs present per case ranged from 0 to 7, with a mean of 1.8 DAVs per case for the entire cohort. DLBCLs displayed the most DAVs per case (mean = 2.2), with PMBLs showing the greatest number of DAVs for any specific diagnostic category (mean = 2.6). Two cases of DLBCL displayed the greatest number of DAVs (7) of any of the cases in the study (Fig. 3). The distributions of DAVs present in the most common large B-cell lymphoma categories and the most common small B-cell lymphoma subcategories are detailed in Figs. 4 and 5, respectively. The types of genetic alterations demonstrated across all cases with DAVs are detailed in Fig. 6.

MPS lymphoma panel oncoprint of all analyzed cases with DAVs detected. TP53 missense mutations were most frequent. DLBCL was the subcategory to have DAVs most frequently identified as well as the greatest DAVs detected per case. The total number of DAVs per case is represented at the top. A limited classification scheme of genetic alteration type is also displayed. Detailed categorization of the consequence of mutations is presented in Fig. 6. DLBCL diffuse large B-cell lymphoma, CLL chronic lymphocytic leukemia, MZL marginal zone lymphoma, FL follicular lymphoma, MCL mantle cell lymphoma, HGBL high grade B-cell lymphoma, LPL lymphoplasmacytic lymphoma, IDA immunodeficiency associated, NOS not otherwise specified.

The four most frequent large B-cell lymphoma categories are shown with the frequency of DAVs in each gene per category represented as a percentage of the total cases in each category. DAVs in TP53 were the most frequent mutation in both DLBCL, GCB (33% of cases) and in DLBCL, ABC (27% of cases). MYD88 was the gene with most frequent DAVs in cases of PCNSL (62.5%), while PMBL cases most frequently had DAVs identified in SOCS1 (45.5%), TNFAIP3 (36.4%), NFKBIE and B2M (27.3% each), and GNA13 (18.2%).

The five most frequent small B-cell lymphoma categories are shown with the frequency of DAVs in each gene per category represented as a percentage of the total cases per category. 10 of the 12 cases of lymphoplasmacytic lymphoma had a mutation in MYD88 (83%), while one-third had DAVs of CXCR4. The most frequent DAVs in cases of MCL were TP53 (18%), ATM (14%), and BIRC3 (7%), while the most frequent DAVs in cases of follicular lymphoma were CREBBP (28%), EZH2 (21%), and TNFRSF14 (19%). Cases of marginal zone lymphoma most frequently had DAVs in TNFAIP3 (15%) and NOTCH2 (12%).

A total of 441 DAVs were identified in the entire cohort of patient samples. The types of genetic alteration are represented here as a percentage of the total DAVs identified. The most frequent type of genetic alterations was missense mutations (45.1%).
Supplementary Tables 1 and 2 list the specific protein, DNA changes, and variant allele frequencies for all DAVs and VUSs observed across the entire cohort, respectively. Overall, 16 variants were ultimately reported as “indeterminate” with respect to VAF (1.8% of total), including 10 DAVs and 6 VUSs, due to a VAF below the validated range of the assay, pool bias, or complex mapping, and thus no VAF was reported.
Overall, 146 cases yielded normal sequencing studies. This group included 49 cases that had no morphologic or immunophenotypic evidence of an MLN. Thus, there were 97 cases with detectable MLN with normal MPS results, of which many had less than 25% tumor cellularity. This observation was further investigated by a second logistic regression analysis to evaluate the effect of preanalytic variables on potential “false negative” MPS results (Table 4). This analysis assumed that certain preanalytic variables, such as tumor percent, may affect the likelihood of obtaining a potential “false negative” MPS result. We defined a potential “false negative” as a case with a detectable MLN but with NV MPS results. We compared these cases to those with positive MPS results (DAV and VUS only) and detectable MLN present. Fresh FNA specimens were excluded from this analysis due to perfect separation with all cases being MPS positive. This analysis revealed that cases with 1–9% tumor (odds ratio 6.38, p value 0.0003) and 10–25% tumor (odds ratio 2.08, p value 0.0454) were significantly more likely to result in NV MPS results compared to cases with ≥50% tumor (as the reference category), indicating a greater potential “false negative” rate among cases with <25% tumor. Although we found no difference between extramural and intramural samples on initial review (not shown in Table 4), fixed bone marrow aspirate clots were all collected at extramural institutions and were significantly more likely to result in NV MPS results compared with other specimen types and sources, indicating that for some specimen types, procedures at the collecting and processing institution may impact the outcome of MPS.
Diagnostic utility of MPS
The detection of DAVs affected the diagnosis in 5.5% (26 of 471) of all analyzed cases (Table 5). Of these, most (21/26, 80.8%) resulted in a refined diagnosis, while the diagnosis was either facilitated (3/26, 11.5%) or changed (2/26, 7.7%) in the remainder. When evaluated based upon only assessing cases with DAVs, there was a diagnostic impact in 10.8% (26/241) of cases. The scenario that was most likely to have a diagnosis refined by MPS results were those with a differential diagnosis of marginal zone lymphoma vs. LPL, which accounted for 34.6% of these cases (9/26).
Discussion
This largest to date and most comprehensive (to the best of our knowledge) study of a targeted MLN MPS panel in routine clinical practice confirms much of what is reported in the scientific literature. While our study demonstrates the modest and somewhat limited current diagnostic impact of MPS in most cases, we identified a subset of cases, albeit minor, in which the sequencing studies affected the final diagnosis. We also identified preanalytic variables that can impact the outcome of sequencing studies, highlighting the importance of appropriate case selection and specimen triage to enhance their more judicious use in routine clinical practice.
In this analysis of our institution’s year-long experience with a targeted MLN MPS panel, diagnostically modifying results were achieved in 5.5% of the total cases analyzed and 10.8% of those cases with DAVs. Refinements of diagnoses were most frequently reported due to either the presence or absence of MYD88 mutations in distinguishing LPL from MZL or the presence of NOTCH2 and TNFAIP3 mutations in favoring a diagnosis of MZL over LPL. Since these mutations are not 100% specific, with, for example, MYD88 mutations also described in other small B-cell neoplasms, these refinements were made in the context of additional laboratory and hematopathologic findings. Other cases which had diagnoses refined by MPS results included those with a differential including T-LGL leukemia vs. other lymphoproliferative disorders or reactive process in which the presence of a STAT3 mutation favored a neoplastic diagnosis. In one of these cases, a novel and heretofore unreported mutation in POT1 led to a diagnosis of T-LGL leukemia [30]. Of note, this POT1 mutation was not detected on the MLN panel; it was identified on another panel performed in tandem in this case that covered different regions of this gene. One difficult case of a PMBL that had a differential diagnosis, including T-cell histiocyte-rich large B-cell lymphoma (THRLBL), was ultimately favored to be PMBL due to the presence of mutations enriched in this entity including GNA13, TNFAIP3, and XPO1 and absence of mutations recurrent in THRLBL such as CREBBP and ATM [31]. Another case, clinically concerning for metastatic squamous cell carcinoma presenting in the humerus had limited material and was originally described as a CD5− CD10− mature B-cell neoplasm. The specimen in this case was not submitted for flow cytometry and had been decalcified, which likely accounted for the inability to detect annexin A1 and BRAF V600E expression by immunohistochemistry. MPS was performed on clotted blood from the biopsy and the case was subsequently classified as HCL based upon the detection of a BRAF V600E mutation, noting that the mutation is not 100% specific for HCL. A plasmablastic neoplasm was ultimately diagnosed as plasmablastic lymphoma rather than plasmablastic myeloma due to the presence of a NOTCH1 mutation. A case of FL, which had a differential including MZL, harbored mutations of EZH2 and TNFRSF14 that supported the diagnosis of FL. A few other cases with refined differential diagnoses ultimately remained descriptive, including cases classified as atypical lymphoproliferations, wherein the presence of clonal DAVs supported neoplastic lymphoid processes over reactive conditions. These determinations, though not definitively diagnostic, led to greater confidence in recommending close clinical follow-up and heightened surveillance.
In addition to the cases which had differential diagnoses refined by the results of a MPS study, there were a few cases in which the final diagnosis was facilitated by MPS results. For example, a noteworthy case from the lymphoma NOS category was that of B-lymphoblastic leukemia/lymphoma arising in a patient with FL. It was unclear if this reflected an unusual transformation of an FL to an aggressive B-cell lymphoma vs. a de novo unrelated lymphoblastic neoplasm; the presence of EZH2 and TNFRSF14 mutations supported the former consideration. Another case, of a limited sample with abundant necrosis, yielded a diagnosis favoring DLBCL, NOS due to the presence of a NOTCH2 mutation. MPS results facilitated the final diagnosis in a case of a plasmablastic lymphoma that had a TP53 mutation supporting transformation from the previously diagnosed CLL (rather than a de novo plasmablastic lymphoma) due to the same DNA change present in both processes.
Two cases underwent a change of diagnosis based on MPS results. One had an initial descriptive diagnosis of “atypical lymphoid infiltrate” and differential considering a T-cell neoplasm with T-follicular helper cell phenotype such as AITL. However, the case did not harbor the canonical mutations characteristic of this lymphoma (IDH2, TET2, and RHOA), but rather displayed a mutation in NOTCH2 supporting a final revised diagnosis of MZL (in the setting of subsequent polyclonal TRG rearrangement studies and monoclonal IGH rearrangement) with the initially concerning T-cell infiltrate with THF-phenotype considered to be reactive. The other case was diagnosed as LPL in the pre-MYD88 era but this was subsequently revised by a different pathologist to MZL, also prior to the advent of MYD88 testing; a later biopsy identified both an MYD88 and CXCR4 mutation, reinstating the initial correct diagnosis.
In addition to the diagnostically useful DAVs identified in our cohort, there were a few cases in which resultant VUSs in isolation were suggested (in addenda by the reviewing hematopathologist) to bear some value in supporting the consideration of a neoplastic infiltrate over a reactive one. In these difficult cases involved by atypical lymphoid infiltrates, VUSs found in isolation may offer some support for a neoplastic process over a reactive process, however, making this determination would rely on detailed and extensive variant review to ensure that the variants identified did not represent benign single nucleotide polymorphisms.
Two of the DLBCL cases had somewhat unusual mutations. One had an SF3B1 mutation and the other a CXCR4 mutation. However, both were explicable from a review of the EMR that revealed that the patients had histories of CLL and LPL, respectively, with these DLBCLs reflecting transformations from these indolent B-cell neoplasms.
Overall, the genetic alterations demonstrated within our cohort are mostly concordant with what has been described in the literature. Our DLBCL cases had the most mutations on average per case, which were often consistent with mutational profiles and clusters recently reported in the literature including frequent mutations in TP53, TNFAIP3, and B2M, as well as frequent mutations in EZH2, TNFRSF14, and CREBBP, and also common MYD88 mutations [1, 14, 17, 32]. Frequently reported mutations in PMBL (SOCS1, TNFAIP3, NFKBIE, GNA13, B2M, and XPO1) and primary CNS DLBCL (MYD88, NOTCH1, and TP53) were observed in our cohort of cases [1, 13, 14, 33,34,35,36]. Other highly enriched mutations that have been described in the mature B-cell lymphomas were present in our study. The cases of FL that we observed commonly displayed TNFRSF14, EZH2, and CREBBP mutations, amongst others [11, 37,38,39,40,41,42,43]. Cases of MCL demonstrated mutations in ATM, TP53, BIRC3, and NOTCH2 [1, 10, 11, 42, 44]. Mutations frequently reported in CLL/SLL including ATM, BIRC3, NOTCH1, SF3B1, and TP53 were common in our cohort [1, 11, 42, 45,46,47,48,49,50,51,52]. Of note, the frequency of TP53 mutations in our cases of CLL/SLL (20%) was moderately higher than that reported in the literature (5–15%) which we infer is likely due to enrichment of relapsed/refractory cases at our institution as a tertiary referral center [11]. This is also likely the explanation for the mildly increased proportion of DLBCL cases with TP53 mutations in our cohort (24%) vs. reported ranges (~10–22%) [53, 54].
In addition, NOTCH2, TNFAIP3, and TNFRSF14 mutations were observed in the marginal zone lymphoma cases, as well as a CREBBP mutation in a case presenting in the lacrimal gland, which is consistent with reports of CREBBP mutations being enriched in ocular adnexal extranodal MZL [1, 11, 42, 55,56,57,58,59,60,61]. Mutations in MYD88 were observed in 83% of our cases of LPL, BRAF mutations were seen in the HCL cases, and a case of HCL variant harbored a MAP2K1 mutation [8, 9, 11, 62,63,64]. In addition, TP53 and NOTCH1 mutations were often observed in the cases of plasmablastic lymphoma and a case of high grade B-cell lymphoma, NOS demonstrated an ID3 mutation [65,66,67]. There were also TP53, TET2, CREBBP, and TNFAIP3 mutations evident in our cases of immunodeficiency associated lymphomas [68, 69]. Of note, an IDH2 mutation was identified in one plasma cell neoplasm (PCN). IDH2 is well recognized as being recurrently mutated in a number of hematologic neoplasms, such as AITL and acute myeloid leukemia, and is not typically associated with PCNs; however, IDH2 mutations have been described to rarely occur in these neoplasms as well [70, 71].
Regarding T-cell neoplasms, STAT3 mutations were often disease defining for our cases of T-LGL [1, 15, 72]. Other common mutations which have been described in T-cell neoplasms were also observed in our cases including TET2 and RHOA mutations in those of TFH origin, TP53, JAK3, and STAT5B mutations in cutaneous T-cell lymphomas, and JAK3 and TP53 mutations in T-prolymphocytic leukemia, amongst others [1, 16, 72,73,74,75,76,77,78,79,80,81,82,83,84].
An additional interesting observation was the presence of mutations in SF3B1 in three bone marrow cases which otherwise had no MLN present. All had prolonged cytopenias leading to bone marrow evaluation, with the differential clinical diagnosis including marrow infiltration by an MLN. In one of these cases, the SF3B1 mutation had a VAF of <5% and can be considered as donor derived clonal hematopoiesis of indeterminate potential in a posttransplant bone marrow from a patient with history of B-lymphoblastic leukemia showing 100% chimerism. The other two cases were diagnosed as myelodysplastic syndrome. Both showed unilineage dysplasia with SF3B1 mutation VAFs of 14% and 25%, respectively. Neither case had prior therapy. These findings suggest a putative unanticipated role for this panel in identifying myeloid neoplasms as having additional value to the interrogation of lymphoid neoplasms.
Another product of our review was the identification of the need for appropriate triage by pathologists of specimens for MPS studies to ensure optimal test utilization practices. Triage items include (1) determination of specimen quality and adequacy for testing, (2) disease state (namely is there actually evidence of disease in the submitted specimen), and (3) test intent (that is for diagnosis, prognosis or prediction). Our analysis of the effect of preanalytic variables (specimen characteristics, fixation, tumor percentage) on MPS processing success and potential “false negative” results showed that extramural cases, which were exclusively fixed, were significantly more likely to fail MPS. Although not specifically evaluated, potential explanations for MPS failure in extramural cases might include institutional differences in specimen collection, fixation processing, and storage procedures. Cases with low tumor percentage were more likely to result in both potential “false negative” and failed MPS. These findings are corroborated by the criteria for acceptable specimens of ≥10% tumor nuclei; however, we also found the rate of potential “false negative” to be enriched in cases with 10–25% tumor, suggesting overall superior diagnostic yield in cases with ≥25% tumor. It is unclear why cases with low tumor percentage resulted with MPS failure. This may be due to overall poorer specimen quality and generally lower specimen cellularity, since this cannot be explained by the lower tumor percentage per se. Unfortunately, overall specimen cellularity was not consistently reported for most of the specimen types included in this analysis, and therefore it is not possible to deduce the fidelity of the denominator in the calculation (i.e., overall specimen cellularity). In addition, the cohort of cases comprising the low tumor percent category was enriched for specimen types with the highest failure rate, including extramural specimens and aspirate clots, which likely contributed to this observation. Fresh samples in this cohort were more likely to have lower tumor percentage, which increased the risk of NV MPS results (a potential “false negative”), but not MPS failure. Conversely, when compared to fresh cases, fixed cases were more likely to fail MPS, but less likely to result in potentially “false negative” MPS results, which is likely due to a relatively higher tumor percentage in the fixed cases. To further illustrate the effect of low tumor percent on “false negative” rate, it is worth noting that of the three cases of THRLBL in this study two showed NV results (the third displayed a VUS in ATM). While this supports our observation that cases with lower tumor percentage are likely to show NV results, it is difficult to draw definitive conclusions since these THRLBLs comprised less than 3% of all DLBCLs. If a specimen is determined to be technically inadequate or to have the potential for spurious results by triage based on the preanalytic variables described here, recommendations against submission for sequencing and for procurement of additional tissue should be provided if possible.
A total of 61 cases sequenced (13% of total) had no evidence of disease, the majority of which showed no detectable mutations (80%), suggesting limited utility of performing this assay when there is no morphologic or immunophenotypic evidence of disease. These inappropriately sequenced negative specimens were often bone marrow aspirates submitted for staging or evaluation of residual disease post treatment and/or pre-transplant, and also peripheral blood specimens with no identifiable evidence of circulating disease. This sequencing panel was not validated for minimal/measurable residual disease detection and therefore this was not an appropriate use of testing. This practice is unnecessary and appropriate triage of these specimens should eliminate the unnecessary cost while improving testing outcomes.
A number of limitations are worth noting. This study was limited by the MPS panel design which included only 40 genes thought to be the most clinically and diagnostically useful regarding association with MLNs at the time this panel was developed. Subsequent to the initial implementation of this panel, further insights into the biology of MLNs have implicated additional genes which may need to be incorporated for more comprehensive coverage and further improvement in diagnostic utility. Additional technical limitations include an inability of the panel to detect large indels, provide information regarding deep intronic splice variants, promoter variants, structural rearrangements, and methylation status data. Another limitation is one of ascertainment bias that is inherent in a retrospective study such as this, in which there was almost certainly biased selection by individual clinicians and/or hematopathologists in determining which cases to submit for MPS. Nevertheless, the fact that we were able to perform MPS on ~50% of unique patients encountered clinically during this year-long study does lend some credibility to its representative nature. The determination of diagnostic utility was largely based on the practice of addenda reporting by the hematopathologist responsible for each case and the respective description of potential utility of MPS results in informing or modifying the original diagnosis. Therefore, an immeasurable degree of variability in practice is inherent in these results. However, detailed chart and report review of all cases in this analysis, including those without addenda, is likely to have minimized this source of bias.
It is imperative to note that our study was focused purely on diagnostic issues as they relate to this MPS panel. It was not an objective of this study to evaluate the wealth of prognostic, predictive, and potential therapeutic information that perhaps exists within these data. These topics will be addressed in a separate study.
While this manuscript was in its final phase of completion, a Swiss group reported their 3-year experience with a 68-gene lymphoma MPS panel applied to 80 cases [27]. They reported that their analysis was useful in most cases, helping to confirm or support diagnoses in 35 of 50 histologically difficult cases. However, while it may appear that their diagnostic utility rate is much higher than ours (70% vs. 5.5%), it should be noted that their cases were highly selected, based upon a predetermined perceived need for mutational analysis, resulting in only ~1% of all cases being analyzed, while our study assessed cases more broadly, agnostic to the anticipated diagnostic value. Nevertheless, their findings complement ours in terms of highlighting the need for appropriate triage and selection of cases since in only a minor proportion will such testing be diagnostically useful.
In conclusion, while this sizable clinical study confirms much of what has been reported in the scientific literature, we have shown that the incorporation of MPS data into the routine comprehensive characterization of MLN cases is not of current diagnostic value in most cases. Improved diagnostic outcomes that led to changing, refining, and facilitating diagnoses were evident in only 5.5% of cases when such testing was applied empirically. Nevertheless, there remains the potential for better outcomes if the practice is standardized. Based upon our findings, a general set of guidelines relating strictly to the diagnostic utility of a panel such as this may emerge. Scenarios in which testing may be particularly useful include (1) a limited sample or an atypical lymphoid infiltrate with equivocal results and when obtaining additional material may be difficult; (2) when a lymphoma is diagnosed, but the classification is ambiguous and a specific genomic profile may help to refine the differential diagnosis; (3) to determine a relationship to a previously diagnosed lymphoma when transformation is suspected; and (4) for the review of archival material, when newly described genetic alterations may alter a previously rendered diagnosis. However, it is important to emphasize that these guidelines for diagnostic use do not address the potential predictive, prognostic, and therapeutically relevant indications for a panel such as this, which were not analyzed as part of this study, in addition to the potential utility in determining eligibility for clinical trials.
Constant review of published literature defining the mutational landscapes of MLNs will be necessary for further refinement in the application of a targeted MLN MPS panel and incorporation of new genes with diagnostic, prognostic, and therapeutic relevance. Thus, while not currently diagnostically essential in the vast majority of cases, the routine role of MPS in MLNs may change in the future with the evolution of novel diagnostic and treatment paradigms. Furthermore, the specific diagnostic categories and timepoints when targeted MLN MPS testing should be routinely performed remain undefined. Many of the mutations in our cases are likely to have more relevance to prognosis and therapy than to diagnosis, which needs to be considered in the value of testing, while some may determine patient eligibility for enrollment in clinical trials. Throughout, it is the responsibility of the pathologist to ensure quality results by exercising strict triage of specimens prior to submission for sequencing including evaluation of specimen quality, disease status, and intent of testing. If implemented effectively, general guidelines for targeted MPS of MLNs that may emerge from studies such as this will provide long-term genetic characterization over the disease course, which will improve diagnostic confidence, inform prognosis, and enhance clinical management.
Change history
22 December 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41379-020-00733-2
References
Swerdlow SH. WHO classification of tumours of haematopoietic and lymphoid tissues. Lyon: International Agency for Research on Cancer; 2017.
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson AG, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–74.
Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–52.
Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33:299–312.
Palumbo GA, Stella S, Pennisi MS, Pirosa C, Fermo E, Fabris S, et al. The role of new technologies in myeloproliferative neoplasms. Front Oncol. 2019;9:321.
Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal hematopoiesis to secondary leukemia. Nat Rev Cancer. 2016;17:5–19.
Malcovati L, Karimi M, Papaemmanuil E, Ambaglio I, Jädersten M, Jansson M, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126:233–41.
Treon SP, Xu L, Yang G, Zhou Y, Liu X, Cao Y, et al. MYD88 L265P somatic mutation in waldenström’s macroglobulinemia. N Engl J Med. 2012;367:826–33.
Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–15.
Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, et al. The 2016 revision of the world health organization classification of lymphoid neoplasms. Blood. 2016;127:2375–90.
Bogusz AM, Bagg A. Genetic aberrations in small B-cell lymphomas and leukemias: molecular pathology, clinical relevance and therapeutic targets. Leuk Lymphoma. 2016;57:1991–2013.
Rosenthal A, Rimsza L. Genomics of aggressive B-cell lymphoma. Hematol Am Soc Hematol Educ Progr. 2018;2018:69–74.
Fukumura K, Kawazu M, Kojima S, Ueno T, Sai E, Soda M, et al. Genomic characterization of primary central nervous system lymphoma. Acta Neuropathol. 2016;131:865–75.
Dubois S, Viailly P, Mareschal S, Bohers E, Bertrand P, Ruminy P, et al. Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: a LYSA study. Clin Cancer Res. 2016;22:2919–28.
Koskela HLM, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmäki H, Andersson EI, et al. Somatic STAT3 mutations in large granular lymphocytic leukemia. N Engl J Med. 2012;366:1905–13.
Wang M, Zhang S, Chuang S, Ashton-Key M, Ochoa E, Bolli N, et al. Angioimmunoblastic T cell lymphoma: novel molecular insights by mutation profiling. Oncotarget. 2017;8:17763–70.
Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24:679–90.
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378:1396–407.
Batlevi C, Shah G, Forlenza C, Intlekofer A. Using genomic data for selecting the treatment of lymphoma patients. Curr Opin Hematol. 2019;26:303–12.
Moffitt AB, Dave SS. Clinical applications of the genomic landscape of aggressive non-Hodgkin lymphoma. J Clin Oncol. 2017;35:955–62.
Pastore A, Jurinovic V, Kridel R, Hoster E, Staiger AM, Szczepanowski M, et al. Integration of gene mutations in risk prognostication for patients receiving first-line immunochemotherapy for follicular lymphoma: a retrospective analysis of a prospective clinical trial and validation in a population-based registry. Lancet Oncol. 2015;16:1111–22.
Qin D. Next-generation sequencing and its clinical application. Cancer Biol Med. 2019;16:4–10.
Kuo FC, Mar BG, Lindsley RC, Lindeman NI. The relative utilities of genome-wide, gene panel, and individual gene sequencing in clinical practice. Blood. 2017;130:433–9.
Kuo FC. Next generation sequencing in hematolymphoid neoplasia. Semin Hematol. 2019;56:2–6.
Galanina N, Bejar R, Choi M, Goodman A, Wieduwilt M, Mulroney C, et al. Comprehensive genomic profiling reveals diverse but actionable molecular portfolios across hematologic malignancies: Implications for next generation clinical trials. Cancers. 2018;11:11.
Surrey LF, MacFarland SP, Chang F, Cao K, Rathi KS, Akgumus GT, et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome Med. 2019;11:32.
Pillonel V, Juskevicius D, Bihl M, Stenner F, Halter JP, Dirnhofer S, et al. Routine next generation sequencing of lymphoid malignancies: clinical utility and challenges from a 3-year practical experience. Leuk Lymphoma. 2020;61:2568–83.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Moosic KB, Paila U, Olson KC, Dziewulska K, Wang TT, Xing JC, et al. Genomics of LGL leukemia and select other rare leukemia/lymphomas. Best Pr Res Clin Haematol. 2019;32:196–206.
Schuhmacher B, Bein J, Rausch T, Benes V, Tousseyn T, Vornanen M, et al. JUNB, DUSP2, SGK1, SOCS1 and CREBBP are frequently mutated in T-cell/histiocyte-rich large B-cell lymphoma. Haematologica. 2019;104:330–7.
Lacy SE, Barrans SL, Beer P, Painter D, Smith A, Roman E, et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a haematological malignancy research network report. Blood. 2020;135:1759–71.
Mansouri L, Noerenberg D, Young E, Mylonas E, Abdulla M, Frick M, et al. Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B-cell lymphoma. Blood. 2016;128:2666–70.
Mottok A, Hung SS, Chavez EA, Woolcock B, Telenius A, Chong LC, et al. Integrative genomic analysis identifies key pathogenic mechanisms in primary mediastinal large B-cell lymphoma. Blood. 2019;134:802–13.
Zhou Y, Liu W, Xu Z, Zhu H, Xiao D, Su W, et al. Analysis of genomic alteration in primary central nervous system lymphoma and the expression of some related genes. Neoplasia. 2018;20:1059–69.
Balint MT, Jelicic J, Mihaljevic B, Kostic J, Stanic B, Balint B, et al. Gene mutation profiles in primary diffuse large B cell lymphoma of central nervous system: next generation sequencing analyses. Int J Mol Sci. 2016;17:683.
Grønbaek K, Worm J, Ralfkiaer E, Ahrenkiel V, Hokland P, Guldberg P. ATM mutations are associated with inactivation of the ARF-TP53 tumor suppressor pathway in diffuse large B-cell lymphoma. Blood. 2002;100:1430.
Gumy-Pause F, Wacker P, Sappino A. ATM gene and lymphoid malignancies. Leukemia. 2004;18:238–42.
Okosun J, Bödör C, Wang J, Araf S, Yang C, Pan C, et al. Integrated genomic analysis identifies recurrent mutations and evolution patterns driving the initiation and progression of follicular lymphoma. Nat Genet. 2014;46:176–81.
Green MR. Chromatin modifying gene mutations in follicular lymphoma. Blood. 2018;131:595–604.
Pasqualucci L, Khiabanian H, Fangazio M, Vasishtha M, Messina M, Holmes AB, et al. Genetics of follicular lymphoma transformation. Cell Rep. 2014;6:130–40.
Mansouri A, Sutton L, Ljungström V, Bondza S, Arngården L, Bhoi S, et al. Functional loss of IκBε leads to NF-κB deregulation in aggressive chronic lymphocytic leukemia. J Exp Med. 2015;212:833–43.
Launay E, Pangault C, Bertrand P, Jardin F, Lamy T, Tilly H, et al. High rate of TNFRSF14 gene alterations related to 1p36 region in de novo follicular lymphoma and impact on prognosis. Leukemia. 2012;26:559–62.
Beà Sílvia, Valdés-Mas Rafael, Navarro Alba, Salaverria Itziar, Martín-Garcia David, Jares Pedro, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110:18250–5.
Giménez N, Martínez-Trillos A, Montraveta A, Lopez-Guerra M, Rosich L, Nadeu F, et al. Mutations in the RAS-BRAF-MAPK-ERK pathway define a specific subgroup of patients with adverse clinical features and provide new therapeutic options in chronic lymphocytic leukemia. Haematologica. 2019;104:576–86.
Quinquenel A, Fornecker L, Letestu R, Ysebaert L, Fleury C, Lazarian G, et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: a FILO group study. Blood. 2019;134:641–4.
Woyach JA, Ruppert AS, Guinn D, Lehman A, Blachly JS, Lozanski A, et al. BTKC481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J Clin Oncol. 2017;35:1437–43.
Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526:519–24.
Landau DA, Tausch E, Taylor-Weiner AN, Stewart C, Reiter JG, Bahlo J, et al. Mutations driving CLL and their evolution in progression and relapse. Nature. 2015;526:525–30.
Ljungström V, Cortese D, Young E, Pandzic T, Mansouri A, Plevova K, et al. Whole-exome sequencing in relapsing chronic lymphocytic leukemia: clinical impact of recurrent RPS15 mutations. Blood. 2016;127:1007–16.
Hernández-Sánchez M, Rodríguez AE, Kohlmann A, Benito R, García JL, Risueño A, et al. TET2 overexpression in chronic lymphocytic leukemia is unrelated to the presence of TET2 variations. Biomed Res Int. 2014;2014:814294.
Puente XS, Pinyol M, Quesada V, Conde L, Ordóñez GR, Villamor N, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–5.
Reddy A, Zhang J, Davis NS, Moffitt AB, Love CL, Waldrop A, et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell. 2017;171:481–494.e15.
Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci USA. 2012;109:3879–84.
Pillonel V, Juskevicius D, Ng CKY, Bodmer A, Zettl A, Jucker D, et al. High-throughput sequencing of nodal marginal zone lymphomas identifies recurrent BRAF mutations. Leukemia. 2018;32:2412–26.
Jung H, Yoo HY, Lee SH, Shin S, Kim SC, Lee S, et al. The mutational landscape of ocular marginal zone lymphoma identifies frequent alterations in TNFAIP3 followed by mutations in TBL1XR1 and CREBBP. Oncotarget. 2017;8:17038–49.
Shin S, Lee S, Kim H, Ki C, Jung CW, Kim J, et al. BRAF V600E and MAP2K1 mutations in hairy cell leukemia and splenic marginal zone lymphoma cases. Ann Lab Med. 2015;35:257–9.
Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209:1537–51.
Kiel MJ, Velusamy T, Betz BL, Zhao L, Weigelin HG, Chiang MY, et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J Exp Med. 2012;209:1553–65.
Ohgami RS, Ma L, Monabati A, Zehnder JL, Arber DA. STAT3 mutations are present in aggressive B-cell lymphomas including a subset of diffuse large B-cell lymphomas with CD30 expression. Haematologica. 2014;99:e105.
Spina V, Khiabanian H, Messina M, Monti S, Cascione L, Bruscaggin A, et al. The genetics of nodal marginal zone lymphoma. Blood. 2016;128:1362–73.
Schmidt J, Federmann B, Schindler N, Steinhilber J, Bonzheim I, Fend F, et al. MYD88 L265P and CXCR4 mutations in lymphoplasmacytic lymphoma identify cases with high disease activity. Br J Haematol. 2015;169:795–803.
Poulain S, Roumier C, Bertrand E, Renneville A, Caillault-Venet A, Doye E, et al. TP53 mutation and its prognostic significance in waldenstrom’s macroglobulinemia. Clin Cancer Res. 2017;23:6325–35.
Waterfall JJ, Arons E, Walker RL, Pineda M, Roth L, Killian JK, et al. High prevalence of MAP2K1 mutations in variant and IGHV4-34–expressing hairy-cell leukemias. Nat Genet. 2014;46:8–10.
Leeman-Neill R, Raciti P, Hsiao S, Vundavalli M, Alobeid B, Mansukhani M, et al. Molecular characterization of post-transplant plasmablastic lymphomas implicates RAS, TP53, and NOTCH mutations and MYC deregulation in disease pathogenesis. Blood. 2017;130:4014.
Stengel A, Kern W, Meggendorfer M, Haferlach T, Haferlach C. Detailed molecular analysis and evaluation of prognosis in cases with high grade b‐cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements. Br J Haematol. 2019;185:951–4.
Gebauer N, Bernard V, Gebauer W, Thorns C, Feller AC, Merz H. TP53 mutations are frequent events in double-hit B-cell lymphomas with MYC and BCL2 but not MYC and BCL6 translocations. Leuk Lymphoma. 2015;56:179–85.
Menter T, Juskevicius D, Alikian M, Steiger J, Dirnhofer S, Tzankov A, et al. Mutational landscape of b‐cell post‐transplant lymphoproliferative disorders. Br J Haematol. 2017;178:48–56.
Courville EL, Yohe S, Chou D, Nardi V, Lazaryan A, Thakral B, et al. EBV-negative monomorphic B-cell post-transplant lymphoproliferative disorders are pathologically distinct from EBV-positive cases and frequently contain TP53 mutations. Mod Pathol. 2016;29:1200–11.
Pawlyn C, Kaiser MF, Heuck C, Melchor L, Wardell CP, Murison A, et al. The spectrum and clinical impact of epigenetic modifier mutations in myeloma. Clin Cancer Res. 2016;22:5783–94.
Langer C, Ibañez M, Liebisch P, Zenz T, Knop S, Einsele H, et al. IDH1 and IDH2 mutations are not frequent in multiple myeloma. Blood. 2010;116:4992.
Greenplate A, Wang K, Tripathi RM, Palma N, Ali SM, Stephens PJ, et al. Genomic profiling of T-cell neoplasms reveals frequent JAK1 and JAK3 mutations with clonal evasion from targeted therapies. JCO Precis Oncol. 2018;2018:1–16.
Odejide O, Weigert O, Lane AA, Toscano D, Lunning MA, Kopp N, et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014;123:1293–6.
Di Napoli A, Jain P, Duranti E, Margolskee E, Arancio W, Facchetti F, et al. Targeted next generation sequencing of breast implant‐associated anaplastic large cell lymphoma reveals mutations in JAK/STAT signalling pathway genes, TP53 and DNMT3A. Br J Haematol. 2018;180:741–4.
Sandell R, Boddicker R, Feldman A. Genetic landscape and classification of peripheral T cell lymphomas. Curr Oncol Rep. 2017;19:1–10.
Gayden T, Sepulveda FE, Khuong-Quang D, Pratt J, Valera ET, Garrigue A, et al. Germline HAVCR2 mutations altering TIM-3 characterize subcutaneous panniculitis-like T cell lymphomas with hemophagocytic lymphohistiocytic syndrome. Nat Genet. 2018;50:1650–7.
Fernandez-Pol S, Costa HA, Steiner DF, Ma L, Merker JD, Kim YH, et al. High-throughput sequencing of subcutaneous panniculitis-like T-cell lymphoma reveals candidate pathogenic mutations. Appl Immunohistochem Mol Morphol. 2019;27:740–8.
Kiel MJ, Sahasrabuddhe AA, Rolland DCM, Velusamy T, Chung F, Schaller M, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK–STAT pathway in sézary syndrome. Nat Commun. 2015;6:8470.
Gros A, Laharanne E, Vergier M, Prochazkova-Carlotti M, Pham-Ledard A, Bandres T, et al. TP53 alterations in primary and secondary sézary syndrome: a diagnostic tool for the assessment of malignancy in patients with erythroderma. PLoS ONE. 2017;12:e0173171.
Da Silva Almeida AC, Abate F, Khiabanian H, Martinez-Escala E, Guitart J, Tensen CP, et al. The mutational landscape of cutaneous T cell lymphoma and sézary syndrome. Nat Genet. 2015;47:1465–70.
Watatani Y, Sato Y, Miyoshi H, Sakamoto K, Nishida K, Gion Y, et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia. 2019;33:2867–83.
McGirt LY, Jia P, Baerenwald DA, Duszynski RJ, Dahlman KB, Zic JA, et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood. 2015;126:508–19.
Choi J, Goh G, Walradt T, Hong BS, Bunick CG, Chen K, et al. Genomic landscape of cutaneous T cell lymphoma. Nat Genet. 2015;47:1011–9.
Kiel MJ, Velusamy T, Rolland D, Sahasrabuddhe AA, Chung F, Bailey NG, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124:1460–72.
Acknowledgements
We would like to thank Lauren E. Steiner, MS, hematopathology service coordinator, Department of Pathology and Laboratory Medicine, Hospital of the University of Pennsylvania, for her valued assistance in identification of cases.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: there was an error in the affiliations and in figure 3.
Supplementary information
Rights and permissions
About this article

Cite this article
Davis, A.R., Stone, S.L., Oran, A.R. et al. Targeted massively parallel sequencing of mature lymphoid neoplasms: assessment of empirical application and diagnostic utility in routine clinical practice. Mod Pathol 34, 904–921 (2021). https://doi.org/10.1038/s41379-020-00720-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41379-020-00720-7
