
Abstract
Identification of p53-positive cells by immunohistochemistry in bone marrow from primary myelodysplastic syndrome patients correlates with the presence of TP53 mutations and poor prognosis. Mutations in the tumor suppressor gene TP53 are more frequent in therapy-related acute myeloid leukemia and myelodysplastic syndrome than in de novo disease, but the role of p53 immunohistochemistry in the therapy-related setting has not been specifically investigated. We studied p53 protein immunoreactivity in bone marrow biopsies of therapy-related myeloid neoplasms and correlated protein expression with TP53 mutation status, clinicopathologic features and outcome. We first studied 32 patients with therapy-related acute myeloid leukemia and 63 patients with therapy-related myelodysplastic syndrome/chronic myelomonocytic leukemia from one institution and then validated our results in a separate group of 32 patients with therapy-related acute myeloid leukemia and 56 patients with therapy-related myelodysplastic syndrome from a different institution. Strong p53 immunostaining in ≥1% of bone marrow cells was highly predictive of a TP53 gene mutation (P<0.0001) and was strongly associated with a high-risk karyotype (P<0.0001). The presence of ≥1% p53 strongly positive cells was associated with poorer overall and disease-specific survival, particularly in the subset of patients treated with stem-cell transplantation. In a multivariable Cox regression model, the presence of ≥1% p53 strongly expressing cells was an independent prognostic marker for overall survival in both cohorts, with hazard ratios of 3.434 (CI: 1.751–6.735, P<0.0001) and 3.156 (CI: 1.502–6.628, P=0.002). Our data indicate that p53 protein expression, evaluated in bone marrow biopsies by a widely available immunohistochemical method, prognostically stratifies patients with therapy-related myeloid neoplasms independent of other risk factors. p53 immunostaining thus represents an easily applicable method to assess risk in therapy-related acute myeloid leukemia/myelodysplastic syndrome patients.
Similar content being viewed by others


Main
Therapy-related myeloid neoplasms include acute myeloid leukemia, myelodysplastic syndrome, and less commonly myelodysplastic/myeloproliferative neoplasms such as chronic myelomonocytic leukemia. Therapy-related myeloid neoplasms occur as late complications of cytotoxic chemotherapy and/or radiotherapy given to treat malignancies or non-malignant conditions.1, 2 The etiology of therapy-related myeloid neoplasms is thought to be related to the mutagenic effect of cytotoxic therapy, including alkylating agents, ionizing radiation therapy, and topoisomerase inhibitors.1 Typically, therapy-related myeloid neoplasms carry complex and usually unbalanced karyotypic abnormalities, particularly loss of material on the long arm of chromosomes 5 and 7 and rearrangements of the 11q23 region involving the MLL gene.3 The prognosis of therapy-related myeloid neoplasms is poor, likely due at least in part to the high proportion of cases with adverse karyotype.4, 5 In therapy-related myelodysplastic syndrome and oligoblastic therapy-related acute myeloid leukemia, the Revised International Prognostic Scoring System has been shown to correlate with outcome, but patients with therapy-related myelodysplastic syndrome appear to do more poorly than de novo myelodysplastic syndrome within each Revised International Prognostic Scoring System risk-stratum.6, 7 Unlike de novo disease, blast count is not strongly associated with outcome in therapy-related myeloid neoplasms and therapy-related myelodysplastic syndrome and therapy-related acute myeloid leukemia are considered together as one disease entity in the 2008 World Health Organization Classification.8 These data suggest that additional markers are needed to stratify the prognosis of therapy-related myeloid neoplasm patients for risk-adapted therapies.
Several types of gene mutations are observed in acute myeloid leukemia and myelodysplastic syndrome, including those affecting epigenetic DNA modification and RNA splicing as well as inactivating mutations of the TP53 tumor suppressor gene.3, 9 Mutational profiles confer important prognostic information that is independent of other traditional risk factors, including karyotype, blast percentage, and patient clinical characteristics in both acute myeloid leukemia and myelodysplastic syndrome.10, 11, 12, 13, 14, 15, 16, 17, 18 The prognostic significance of mutations in therapy-related myeloid neoplasms has not been extensively characterized. Point mutations of the tumor suppressor gene TP53 represent the most frequent single genetic abnormality in therapy-related myeloid neoplasms and are detected in 20–40% of therapy-related myeloid neoplasm patients compared with 5–20% in de novo acute myeloid leukemia and myelodysplastic syndrome.3, 13, 15, 19 TP53 mutations are often associated with loss of heterozygosity of the TP53 locus and cytogenetic defects such as −5/del(5q) and 17p-.3, 9, 13, 19, 20, 21 The importance of TP53 mutations has been validated in myelodysplastic syndrome in the non-therapy-related setting, particularly in myelodysplastic syndrome with del(5q).21, 22 One recent study showed that TP53 mutations in therapy-related myeloid neoplasms predicted an inferior clinical outcome compared with wild-type TP53, but the authors examined a relatively small number of cases.15
Complete assessment of TP53 mutation requires sequencing of all exons, because mutations often occur outside of the most commonly recognized hot spots, and requires a highly sensitive assay, because the mutation may only be present in a subset of the hematopoietic cells.13, 21 TP53 mutation causes overexpression of the mutated protein and several recent studies have shown that TP53 mutations in solid tumors, lymphomas, and myelodysplastic syndrome can be accurately predicted by immunohistochemistry using widely available antibodies to the p53 protein.21, 23, 24 In this study, we sought to determine the prevalence of p53 protein immunoreactivity in bone marrow samples from therapy-related myeloid neoplasm patients and to evaluate its association with mutations in the TP53 gene. We also sought to assess whether p53 immunoreactivity was associated with patient outcome in two large cohorts of therapy-related myeloid neoplasm patients with clinical follow-up.
Materials and methods
Patient Population and Samples
The cases in the test set were identified by a search of the pathology database of Massachusetts General Hospital for consecutive cases of therapy-related myeloid neoplasms diagnosed between 1/1/2001 and 9/30/2013 in which paraffin blocks from the original diagnostic biopsy were available. A total of 95 therapy-related myeloid neoplasms (60 therapy-related myelodysplastic syndrome, 3 therapy-related chronic myelomonocytic leukemia, and 32 therapy-related acute myeloid leukemia) were found. Original diagnostic bone marrow biopsies from 88 patients with therapy-related myeloid neoplasms (56 therapy-related myelodysplastic syndrome and 32 therapy-related acute myeloid leukemia) diagnosed at the University of Texas MD Anderson Cancer Center between 1/1/2007 and 8/31/2010 comprised the validation set. All patients in both cohorts had a documented history of a benign or malignant condition for which they received radiotherapy alone, chemotherapy alone, or combined chemoradiotherapy prior to the diagnosis of therapy-related myeloid neoplasm. Clinical information and follow-up data were obtained from the medical record. Cytogenetic analysis was performed with trypsin-Giemsa banding techniques on bone marrow cells from aspirates obtained from the diagnostic bone marrow sample and karyotype abnormalities were described according to the International System for Human Cytogenetic Nomenclature. The myelodysplastic syndrome and chronic myelomonocytic leukemia karyotypes were stratified by the Comprehensive Cytogenetic Scoring System of the Revised International Prognostic Scoring System6, 25 and the acute myeloid leukemia karyotypes were stratified by the United Kingdom Medical Research Council system.26 For analyses in which therapy-related myelodysplastic syndrome and therapy-related acute myeloid leukemia were considered together, high-risk karyotype was defined for therapy-related myelodysplastic syndrome/chronic myelomonocytic leukemia as Revised International Prognostic Scoring System high or very high risk and for therapy-related acute myeloid leukemia as United Kingdom Medical Research Council adverse risk. Therapies administered were recorded as supportive care (transfusion support and anti-infectives), low-intensity therapies (low-dose cytotoxic therapies, including hypomethylating agents), induction chemotherapy, or allogeneic stem-cell transplantation. This study was approved by the Institutional Review Boards at both Massachusetts General Hospital and the University of Texas MD Anderson Cancer Center.
Immunohistochemistry for p53
Bone marrow biopsies from Massachusetts General Hospital were fixed in B+ fixative (BBC Biochemical, Seattle, WA, USA) and decalcified in RapidCal Immuno (BBC Biochemical) for 30 min at room temperature prior to routine tissue processing and paraffin-embedding. Bone marrow biopsies from the University of Texas MD Anderson Cancer Center were fixed in neutral-buffered formalin and decalcified in 10% formic acid for 3 h at 50 °C in a microwave oven. Paraffin-embedded tissues sections (3 microns) were stained with p53 monoclonal antibody DO-7 (PA0057 Ready-to-use, Leica Biosystems, Newcastle, UK) in a Leica Bond-III immunostainer according to the manufacturer’s instructions. The DO-7 antibody recognizes both wild-type and mutant p53 protein.
p53 Immunohistochemistry Scoring
The percentage of cells with strong nuclear p53 staining was counted at × 400 magnification on immunostained bone marrow sections by a hematopathologist (RPH) who was blinded to the clinical and outcome information of the cases. Only hematopoietic cells with strong (3+) nuclear staining were counted. At least 300 cells (median 610 in test set and 576 in validation set) were counted except in cases with >15% p53 3+ cells present throughout the biopsy and in one paucicellular case in which no p53 3+ cells were identified in all 241 mononuclear cells available. On the basis of prior studies,22 a cutoff of ≥1% p53 3+ cells was used to define positive p53 staining (p53 immunohistochemistry-positive).
TP53 Sequencing
Mutation analyses of the TP53 gene was performed in 71 patients using DNA from archived fixed and frozen cell pellets from the original diagnostic bone marrow sample, using the clinically validated Snapshot-NGS-V1 assay to detect single nucleotide variants and insertions/deletions. Briefly, DNA was isolated using the QIAmp (Qiagen, Hilden, Germany). Sequencing libraries were made using an anchored multiplex PCR assay and quantified using KAPA qPCR reagents (Kapa Biosystems Inc., Woburn, MA, USA) per the manufacturer’s instructions. All 11 exons of TP53 were amplified (23 amplicons of approximately 100 bp each) and sequenced using Illumina MiSeq (Illumina, Inc., San Diego, CA, USA). Sequence reads were aligned against a hg19 reference genome with an average sequencing coverage of 300x.
Statistical Analysis
Fisher’s exact test, Sperman rank-order correlation coefficient, and Mann-Whitney test were used to evaluate differences between clinicopathologic features and p53 protein expression. Overall survival was defined as time from diagnosis to death from any cause. Disease-specific survival was defined as time from diagnosis to death from myelodysplastic syndrome or acute myeloid leukemia, with patients censored if death was due to other causes (such as complications of another non-myeloid neoplasm). The method of Kaplan-Meier and the log-rank test were used to compare overall survival and disease-specific survival between groups. Independent variables predicting survival were evaluated in a multivariable model using Cox Regression analyses. All two-sided P values of <0.05 were considered statistically significant. Prism 4.0 and XLSTAT were used for data analyses.
Results
Clinical, Pathologic, and Genetic Features of Therapy-Related Myeloid Neoplasm Patient Groups
The characteristics of test set and validation set are shown in Table 1. Compared with the test set population, the validation set patients were younger (P=0.03), included fewer patients treated with radiotherapy only (P=0.0001), and were treated less often with supportive care only (P=0.03). The World Health Organization disease subtypes were similar between the test and validation sets. Although the myelodysplastic syndrome patients in the validation set included a higher proportion of patients with higher-risk karyotypes (P=0.001), there was no significant difference in the overall Revised International Prognostic Scoring System risk scores between the test and validation set myelodysplastic syndrome patients (Table 1). The primary diseases for which patients in the test and validation sets received cytotoxic therapy are listed in Supplementary Table 1.
Results of p53 Immunostaining in the Test Set
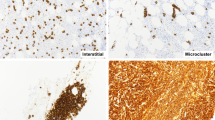
Strong p53-expressing cells (3+) were observed in 67/95 (71%) of the biopsies, ranging from 0.1 to 57.5% of all nucleated bone marrow hematopoietic cells. Using a ≥1% cutoff of p53 3+ cells (defined as p53 immunohistochemistry-positive), 40/95 (42%) of the samples, including 10/32 therapy-related acute myeloid leukemia (31%) and 30/63 therapy-related myelodysplastic syndrome/chronic myelomonocytic leukemia (48%), were p53 immunohistochemistry-positive. A detailed distribution of the percentage of p53 immunohistochemistry 3+ cell percentages in the test set is shown in Supplementary Table 2. The median percentage of p53 3+ cells among the p53 immunohistochemistry-positive cases was 16.9% (range 1.1–57.5%). The cells staining for p53 were mostly early erythroid forms, blasts, and early myeloid cells that lacked nuclear segmentation (Figure 1). Variable staining was also seen in megakaryocytes. Overall, 3+ megakaryocyte staining was present in 16/89 (18%) cases with evaluable megakaryocytes and in 15 of these 16 cases (94%), ≥1% p53 3+ cells were also present. In one case with 3+ megakaryocyte staining, only 0.5% of the total cells showed 3+ p53 staining.

Representative images of p53 immunostaining in bone marrow biopsies from patients with therapy-related myeloid neoplasms. (a) Case of therapy-related myelodysplastic syndrome (refractory anemia with excess blasts-1) with very rare p53 3+ cells (0.1%). (b) Case of therapy-related myelodysplastic syndrome (refractory anemia with excess blasts-2) with frequent p53 3+ cells (10.2%). (c) Case of therapy-related myelodysplastic syndrome (refractory cytopenia with multilineage dysplasia, 2% blasts) with numerous p53 3+ cells (41.3%). (d) Case of therapy-related acute myeloid leukemia with frequent p53 1+ and 2+ cells, but with only rare p53 3+ cells (0.1%). (e) Case of therapy-related acute myeloid leukemia with numerous p53 3+ cells (31.9%). (f) Case of p53 immunohistochemistry-negative therapy-related myelodysplastic syndrome (refractory cytopenia with multilineage dysplasia) with only 0.5% p53 3+ cells and with rare p53 3+ megakaryocytes.
In order to evaluate the reproducibility of p53 scoring, 40 cases (including all 18 cases with 0.5–5% p53 3+ cells) were evaluated independently by two additional observers (VN and AC). With respect to scoring cases as p53 immunohistochemistry-positive (≥1%) or negative (<1%), all three observers agreed on 37/40 cases (93%). For the three discrepant cases, the scores were 0.6-1.0-1.4, 0.0-1.1-1.1, and 0.8-1.1-2.3%. The Spearman r correlation coefficients of p53 3+ percentages between the three observers were 0.95, 0.87, and 0.86 (P<0.0001 for all comparisons) and the kappa coefficients between the three observer pairs with respect to p53 immunohistochemistry-positive vs p53 immunohistochemistry-negative were 0.95, 0.90, and 0.85.
Comparison of features between p53 immunohistochemistry-positive and p53 immunohistochemistry-negative cases in the test set is shown in Table 2. There was no association between p53 immunohistochemistry status and patient age, hemoglobin level, bone marrow cellularity, peripheral blood blast percentage, or diagnosis as therapy-related myelodysplastic syndrome/chronic myelomonocytic leukemia vs therapy-related acute myeloid leukemia. p53 immunohistochemistry-positive status was borderline associated with a lower white blood count, lower platelet count, and lower bone marrow blast percentage. Positivity for p53 was seen less often in patients with prior exposure to radiation therapy only compared with chemotherapy only or combined modality therapy (P=0.02), but there was no difference in latency to develop therapy-related myeloid neoplasms between p53 immunohistochemistry-positive and p53 immunohistochemistry-negative cases (Table 2). p53 immunohistochemistry positivity was also strongly associated with higher karyotype risk in the entire cohort (P<0.0001) and in therapy-related myelodysplastic syndrome/chronic myelomonocytic leukemia (P<0.0001), but was only borderline associated with higher United Kingdom Medical Research Council karyotype risk in therapy-related acute myeloid leukemia (P=0.10). There was a strong association between p53 immunohistochemistry positivity and the presence of −5/del(5q) and −7/del(7q) (both P<0.0001), but there was no significant association with the presence of 17p abnormalities. No cases with 11q23 abnormality showed p53 immunohistochemistry positivity, compared with 13% of the p53 immunohistochemistry-negative cases (P=0.04). p53 immunohistochemistry positivity was also associated with higher overall Revised International Prognostic Scoring System risk grouping in the therapy-related myelodysplastic syndrome/chronic myelomonocytic leukemia patients (P<0.0001). Regarding morphologic features, p53 immunohistochemistry positivity was associated with the presence of marked (>50% of lineage) erythroid dysplasia (P=0.015) and was borderline associated with marked megakaryocytic dysplasia (P=0.057), but was not associated with myeloid lineage dysplasia.
Association of p53 Immunohistochemistry Results with TP53 Mutations
Sequencing of the TP53 gene was informative in 65/71 (92%) of the tested cases. The sequencing results and correlation with p53 immunohistochemistry results are shown in Supplementary Table 3. Among the p53 immunohistochemistry-positive cases, 29/31 (94%) had a TP53 mutation identified, including 23 with single, 5 with two, and 1 with three missense mutations; 2 cases additionally had TP53 insertions/deletions. In both of the p53 immunohistochemistry-positive cases with no TP53 mutation identified, the p53 immunohistochemistry was only borderline positive (1.1% for both). Conversely, 4/34 (12%) p53 immunohistochemistry-negative cases had a TP53 mutation identified. Two of these discrepant cases involved nonsense mutations creating a stop codon (c.310C>T/p.Gln104* and c.586C>T/p.Arg196*), resulting in a truncated protein that would likely not be detected by immunohistochemistry. The other two cases had single missense mutations (c.529C>T/p.Pro177Ser and c.393C>A/p.Asn131Lys) with allele burdens of 29% and 30%, respectively. Overall, p53 immunohistochemistry positivity was strongly associated with the presence of TP53 mutation (P<0.0001). There was no correlation between the percentage of p53 3+ cells and the estimated mutant allele burden by sequence analysis (Spearman r correlation coefficient=0.045, P=0.81).
Association of p53 Immunohistochemistry Status with Patient Outcome in the Test Set
The median follow-up of all patients was 12 months and the median follow-up of surviving patients was 43 months. The median overall survival of all patients was 12 months and the median disease-specific survival was 13 months. In univariate analysis of all patients in the test group, p53 immunohistochemistry-positive status was strongly associated with shorter overall survival (5.2 vs 20.6 months, P<0.0001) and disease-specific survival (5.3 vs 25.4 months, P<0.0001) compared with p53 immunohistochemistry-negative patients (Figures 2a and b). The unfavorable impact of p53 immunohistochemistry-positive status was seen in both therapy-related acute myeloid leukemia patients (median overall and disease-specific survival 4.2 vs 15.9 months, P=0.002) and therapy-related myelodysplastic syndrome/chronic myelomonocytic leukemia patients (median overall survival 5.4 vs 25.4 months, P<0.0001, median disease-specific survival 5.5 vs 36.7 months, P<0.0001) and was maintained in patients with high-risk karyotypes (median overall survival 5.4 vs 19.7 months, P=0.0005, median disease-specific survival 5.5 vs 19.7 months, P=0.0006) (Figure 2c). The survival of patients treated with stem-cell transplantation was also markedly influenced by p53 immunohistochemistry status (median overall survival and disease-specific survival 11.5 months for p53 immunohistochemistry-positive vs 98 months for p53 immunohistochemistry-negative patients, P<0.0001) (Figure 2d).

Outcome of patients with therapy-related myeloid neoplasms (t-MN) in the test and validation groups according to p53 immunohistochemistry status. In the test group, overall survival (a) and disease-specific (b) survival of p53 immunohistochemistry-positive (p53 IHC+) patients (n=40) are shorter than those of p53 immunohistochemistry-negative (p53 IHC−) patients (n=55) (P<0.0001 for both, log rank test). Among therapy-related myeloid neoplasm patients in the test group with high-risk karyotypes (c), p53 immunohistochemistry-positive patients (n=32) have shorter overall survival than p53 immunohistochemistry-negative patients (n=18) (P=0.0005, log rank test). Among therapy-related myeloid neoplasm patients in the test group treated with stem-cell transplantation (d), p53 immunohistochemistry-positive patients (n=9) have shorter overall survival than p53 immunohistochemistry-negative patients (n=16) (P<0.0001, log rank test). In the validation set, overall survival (e) and disease-specific survival (f) of p53 immunohistochemistry-positive patients (n=34) are shorter than those of p53 immunohistochemistry-negative patients (n=50) (P=0.0001 and P<0.0001, respectively, log rank test).
The percentages of p53 2+ and 1+ cells were also scored, but there was no correlation between either p53 1+/2+ cells or the percentage of cells showing any p53 staining (1–3+) with outcome (data not shown). In contrast, the presence of any (1–3+) p53 staining of megakaryocytes was associated with a shorter overall survival (P=0.0001). However, the overall p53 immunohistochemistry status (≥1% 3+ cells among all hematopoietic cells) had a higher hazard ratio for overall survival (HR=5.099, 95% CI 2.906–8.944) compared with any megakaryocyte staining (HR=2.727, 95% CI 1.635–4.547). We investigated whether cutoffs other than ≥1% may better define a prognostically relevant group of therapy-related myeloid neoplasms cases by p53 staining. Compared with alternative cutoffs of 0.1, 0.2, 0.3, 0.5, 0.7, 2, 5, and 10%, the cutoff of ≥1% p53 3+ cells provided the highest hazard ratio with respect to both overall and disease-specific survival (data not shown). With respect to correlation with TP53 mutation status, a cutoff of ≥2% provided more specificity but lower sensitivity: 28/28 (100%) with ≥2% p53 3+ cells vs 29/31 (94%) with ≥1% p53 3+ cells were TP53 mutated, while 5/37 (14%) with <2% p53 3+ cells vs 4/34 (12%) with <1% p53 3+ cells lacked TP53 mutation. Univariate analysis excluding 12 cases with borderline p53 scores (0.6– 2%) showed retained significance of p53 immunohistochemistry positivity for shorter overall and disease-specific survival (P<0.0001 for both).
In a multivariable Cox regression model including p53 immunohistochemistry status, patient age, therapy-related acute myeloid leukemia vs therapy-related myelodysplastic syndrome/chronic myelomonocytic leukemia, high-risk karyotype, and stem-cell transplantation, the p53 immunohistochemistry-positive status was a strong independent predictor of shorter overall survival (HR 3.434, P<0.0001) and disease-specific survival (HR 3.882, P<0.0001) (Table 3).
Association of p53 Immunohistochemistry Status with Patient Outcome in the Validation Set
p53 immunohistochemistry was performed on a separate validation set of 88 therapy-related myeloid neoplasms diagnosed at the University of Texas MD Anderson Cancer Center, and adequate tissue for interpretation was present in 84 of the cases. p53 immunohistochemistry positivity (≥1% cells with 3+ p53 staining) was observed in 34/84 (40%) of the biopsies, including 8/28 therapy-related acute myeloid leukemia (28%) and 26/56 therapy-related myelodysplastic syndrome (46%). A detailed distribution of the percentage of p53 3+ cell percentages in the validation set is shown in Supplementary Table 2. The median percentage of p53 3+ cells among the p53 immunohistochemistry-positive cases was 8.1% (range 1.2–46.7%). The median follow-up time in the validation set of all patients was 10 months and of surviving patients was 56 months. In univariate analysis of the validation set, p53 immunohistochemistry positivity was strongly associated with shorter overall survival (median 7.7 vs 14.6 months, P=0.0001) and disease-specific survival (median 10.1 vs 45.1 months, P<0.0001) (Figures 2e and f). Even after excluding the seven cases with borderline p53 3+ percentages (0.6–2.0%, comprising 8% of the entire validation cohort), p53 immunohistochemistry positivity retained its significance for both overall survival (P=0.0006) and disease-specific survival (P=0.0004).
In a multivariable Cox regression model including the same variables applied to the test set, p53 immunohistochemistry-positive status was a strong independent predictor of shorter overall survival (HR 3.156, P=0.002) and disease-specific survival (HR 2.467, P=0.003) in the validation set (Table 4).
Discussion
Several studies have shown that TP53 mutations in myelodysplastic syndrome predict inferior clinical outcome compared with wild-type TP53.4, 13, 15, 21, 22 To our knowledge, the current study is the first to examine p53 protein expression in a large series of therapy-related myeloid neoplasms. We scored p53 by manual counting and found that, using a cutoff similar to that used in low-risk myelodysplastic syndrome (≥1% 3+ cells),22 positivity for p53 was strongly associated with poor outcome in therapy-related myeloid neoplasms. This adverse association was independent of other factors known to influence prognosis, such as age, acute myeloid leukemia vs myelodysplastic syndrome, karyotype risk group, and treatment with stem-cell transplantation. Importantly, we validated these results in a cohort of therapy-related myeloid neoplasm patients from another institution that displayed different karyotype profiles and treatment backgrounds, likely reflecting different referral patterns and patient populations at the two institutions. The fact that we found similar results in the test and validation cohorts, with strong prognostic significance for p53 immunohistochemistry in both univariate and multivariable analyses in both cohorts, supports the utility of p53 immunohistochemistry to predict prognosis across a wide spectrum of therapy-related myeloid neoplasms in different practice settings.
Prior studies in de novo myelodysplastic syndrome have shown that the presence of TP53 mutations correlates well with p53 protein expression. In a large series of patients with de novo myelodysplastic syndrome, 73% of patients with TP53 mutation showed strong p53 expression by immunohistochemistry, whereas none of the patients with wild-type TP53 showed strong p53 expression.21 In that study, p53 staining was assessed based on a combination of the percentage of cells stained and staining intensity. In patients with low-risk myelodysplastic syndrome with del(5q), it was also shown that the presence of ≥1% p53-expressing cells was positively correlated with TP53 mutations.13, 22 We also found an excellent correlation between≥1% p53 3+ cells and the presence of TP53 mutation in the 71 cases subjected to sequencing in our study: 29/33 cases (88%) with TP53 mutation were scored as p53 immunohistochemistry-positive. The four false-negative cases included two cases with a nonsense mutation truncating the protein, likely explaining the loss of detection by immunohistochemistry.21 Interestingly, the other two cases with TP53 mutation and negative p53 immunohistochemistry occurred in patients with non-complex karyotypes, 45,XY,−7,del(20)(q11.2) and 46,XY,del(7)(q22q31), who had relatively long survivals of 69.1 and 18.8 months, respectively. According to the database of the International Agency for Research on Cancer (http://p53.iarc.fr/TP53GeneVariations.aspx), both of these mutations are predicted to result in a partially functional protein, whereas in 28/29 of the TP53 mutated cases in our series that were p53 immunohistochemistry-positive, the mutations were predicted to yield a non-functional protein. Although no definitive conclusions can be drawn from these two discrepant cases, the findings raise the possibility that missense mutations that do not increase p53 protein expression may reflect less deleterious effects on the protein and result in a disease phenotype distinct from TP53 mutations that result in an overexpressed, non-functional protein.
Many of the cases in our series had frequent cells with weak to moderate p53 immunostaining and they appeared to have no relevance with respect to outcome or correlation with karyotypes (data not shown). Saft et al22 showed by single-cell microdissection that cells with weak to moderate p53 staining lack TP53 mutation. Therefore, in counting p53 immunostained cells, it is important to avoid counting moderately or weakly stained cells. Similar to the Saft et al22 study, we found excellent interobserver concordance in manual enumeration of p53 3+ cells using a cutoff of ≥1%. The Saft et al study additionally found concordance between manual and automated counting and between two alternative p53 antibodies (DO1 and DO7). These results suggest that p53 immunohistochemistry can represent a reliable clinical tool for risk stratification. However, we found some lack of interobserver concordance in cases with p53 immunohistochemistry scores of 1–2% and among the three cases with p53 immunohistochemistry scores of 1–2%, two cases lacked TP53 mutation. Thus, p53 immunohistochemistry scores of 1–2% should be interpreted with caution and may be less reliable in predicting a TP53 mutation than p53 immunohistochemistry scores of >2%. Further study is required on cases with borderline p53 immunohistochemistry scores in order to determine the mutation incidence in this subset.
The precise role of TP53 mutations in the pathogenesis of therapy-related myeloid neoplasms is uncertain. Although TP53 mutations are more frequent in therapy-related myelodysplastic syndrome and therapy-related acute myeloid leukemia compared with their de novo counterparts, it is unclear if this merely reflects the higher incidence of adverse karyotypes that are known to be strongly associated with therapy-related disease. TP53 mutations appear to be an early and relatively stable event in myelodysplastic syndrome, as they have been shown to be present many years before disease progression13 and were not seen to occur as a secondary event after the initial diagnosis in one study that evaluated for the mutation at multiple time points.21 Additionally, low-level TP53 mutations have been found in the blood and bone marrow from patients who subsequently develop therapy-related myeloid neoplasms, even prior to the cytotoxic exposure.27 This has led some authors to suggest that cytotoxic therapy may select for TP53-mutated clones, which subsequently contribute to the complex cytogenetic features and aggressive biologic behavior of therapy-related myeloid neoplasms.27 However, despite the fact that altered TP53 is strongly associated with high-risk karyotypes, 36% of the high-risk karyotype therapy-related myeloid neoplasms in our test set were p53 immunohistochemistry-negative. We also found that p53 immunohistochemistry positivity was less frequent in patients exposed to radiation only, correlating with the observation that therapy-related myeloid neoplasms following radiation only have cytogenetic features and clinical behavior that differ from therapy-related myeloid neoplasms following cytotoxic chemotherapy.28 These data suggest that the molecular mechanisms underlying the etiology of therapy-related myeloid neoplasms are diverse and are likely associated with different disease behaviors.
In our study, p53 immunohistochemistry positivity was strongly correlated with −5/del(5q) cytogenetic abnormalities and usually occurred in the context of a complex karyotype. These results are generally similar to those reported for de novo myelodysplastic syndrome.21, 29 However, we found a higher incidence of p53 immunohistochemistry positivity in cases lacking −5/del(5q) (6/55, 11%) than the 1.5% reported in the recent series of de novo myelodysplastic syndrome.21 Positivity for p53 immunohistochemistry occurred in two patients with normal karyotype in the test group and in two patients with isolated −7/del(7q) in the validation group (data not shown). In myelodysplastic syndrome with del(5q), TP53 mutation and p53 immunopositivity predicts poor response to lenalidomide13, 21 and none of the patients with TP53 mutation showed a complete cytogenetic response to lenalidomide, indicating that information on the p53 status is not only important for prognostic stratification, but may also predict response to particular therapies. In our study, we found that, although the proportions of p53 immunohistochemistry-positive and p53 immunohistochemistry-negative patients treated with stem-cell transplantation were similar, p53 immunohistochemistry-positive therapy-related myeloid neoplasm patients undergoing stem-cell transplantation had a very poor outcome: eight of nine patients died because of disease relapse at a median of 5 months (range 2–10 months) after stem-cell transplantation.
In summary, we found that p53 protein expression, defined as ≥1% cells with strong nuclear staining in bone marrow biopsies, is frequently found in therapy-related myelodysplastic syndrome and therapy-related acute myeloid leukemia and reflects the high incidence of TP53 missense mutations in therapy-related myeloid neoplasms. p53 immunostaining prognostically stratifies therapy-related myeloid neoplasm patients independent of other well-known risk factors and in particular identifies patients with very poor outcome following stem-cell transplantation. In combination with other markers of disease aggressiveness and patient characteristics, p53 immunohistochemistry may help guide the therapeutic approach to this aggressive group of myeloid neoplasms.
References
Bhatia S . Therapy-related myelodysplasia and acute myeloid leukemia. Semin Oncol 2013;40:666–675.
Godley LA, Larson RA . Therapy-related myeloid leukemia. Semin Oncol 2008;35:418–429.
Pedersen-Bjergaard J, Andersen MK, Andersen MT et al. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia 2008;22:240–248.
Christiansen DH, Andersen MK, Pedersen-Bjergaard J . Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol 2001;19:1405–1413.
Bacher U, Haferlach C, Alpermann T et al. Patients with therapy-related myelodysplastic syndromes and acute myeloid leukemia share genetic features but can be separated by blast counts and cytogenetic risk profiles into prognostically relevant subgroups. Leuk Lymphoma 2013;54:639–642.
Greenberg PL, Tuechler H, Schanz J et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood 2012;120:2454–2465.
Ok CY, Hasserjian RP, Fox PS et al. Application of the international prognostic scoring system-revised in therapy-related myelodysplastic syndromes and oligoblastic acute myeloid leukemia. Leukemia 2014;28:185–189.
Singh ZN, Huo D, Anastasi J et al. Therapy-related myelodysplastic syndrome: morphologic subclassification may not be clinically relevant. Am J Clin Pathol 2007;127:197–205.
Pedersen-Bjergaard J, Andersen MT, Andersen MK . Genetic pathways in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Hematology Am Soc Hematol Educ Program 2007;392–397.
Bejar R, Stevenson KE, Caughey BA et al. Validation of a prognostic model and the impact of mutations in patients with lower-risk myelodysplastic syndromes. J Clin Oncol 2012;30:3376–3382.
Cancer Genome Atlas Research N. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med 2013;368:2059–2074.
Grossmann V, Schnittger S, Kohlmann A et al. A novel hierarchical prognostic model of AML solely based on molecular mutations. Blood 2012;120:2963–2972.
Jadersten M, Saft L, Smith A et al. TP53 mutations in low-risk myelodysplastic syndromes with del(5q) predict disease progression. J Clin Oncol 2011;29:1971–1979.
Patel JP, Gonen M, Figueroa ME et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–1089.
Shih AH, Chung SS, Dolezal EK et al. Mutational analysis of therapy-related myelodysplastic syndromes and acute myelogenous leukemia. Haematologica 2013;98:908–912.
Shivarov V, Gueorguieva R, Stoimenov A et al. DNMT3A mutation is a poor prognosis biomarker in AML: results of a meta-analysis of 4500 AML patients. Leuk Res 2013;37:1445–1450.
Thol F, Kade S, Schlarmann C et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012;119:3578–3584.
Walter MJ, Shen D, Shao J et al. Clonal diversity of recurrently mutated genes in myelodysplastic syndromes. Leukemia 2013;27:1275–1282.
Ben-Yehuda D, Krichevsky S, Caspi O et al. Microsatellite instability and p53 mutations in therapy-related leukemia suggest mutator phenotype. Blood 1996;88:4296–4303.
Rucker FG, Schlenk RF, Bullinger L et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012;119:2114–2121.
Kulasekararaj AG, Smith AE, Mian SA et al. TP53 mutations in myelodysplastic syndrome are strongly correlated with aberrations of chromosome 5, and correlate with adverse prognosis. Br J Haematol 2013;160:660–672.
Saft L, Karimi M, Ghaderi M et al. p53 protein expression independently predicts outcome in patients with lower-risk myelodysplastic syndromes with del(5q). Haematologica 2014;99:1041–1049.
Giles FJ, Bekele BN, O'Brien S et al. A prognostic model for survival in chronic lymphocytic leukaemia based on p53 expression. Br J Haematol 2003;121:578–585.
Chang H, Jiang AM, Qi CX . Aberrant nuclear p53 expression predicts hemizygous 17p (TP53) deletion in chronic lymphocytic leukemia. Am J Clin Pathol 2010;133:70–74.
Schanz J, Tuchler H, Sole F et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol 2012;30:820–829.
Grimwade D, Hills RK, Moorman AV et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010;116:354–365.
Wong TN, Ramsingh G, Young A et al. The role of early TP53 mutations on the evolution of therapy-related AML (abstract). Blood 2013;15:5.
Nardi V, Winkfield KM, Ok CY et al. Acute myeloid leukemia and myelodysplastic syndromes after radiation therapy are similar to de novo disease and differ from other therapy-related myeloid neoplasms. J Clin Oncol 2012;30:2340–2347.
Volkert S, Kohlmann A, Schnittger S et al. Association of the type of 5q loss with complex karyotype, clonal evolution, TP53 mutation status, and prognosis in acute myeloid leukemia and myelodysplastic syndrome. Genes Chromosomes Cancer 2014;53:402–410.
Acknowledgements
We would like to thank Julie Batten, Divya Panditi, and Yi Cao for assistance with the sequencing, Hildreth Curran for assistance in formatting the manuscript, and Steve Conley for assistance with formatting the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article

Cite this article
Cleven, A., Nardi, V., Ok, C. et al. High p53 protein expression in therapy-related myeloid neoplasms is associated with adverse karyotype and poor outcome. Mod Pathol 28, 552–563 (2015). https://doi.org/10.1038/modpathol.2014.153
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2014.153
This article is cited by
-
The expression level of ARF and p53 in AML patients, and their relation to patients' outcome
Egyptian Journal of Medical Human Genetics (2023)
-
The International Consensus Classification of myelodysplastic syndromes and related entities
Virchows Archiv (2023)
-
Characteristics and outcomes of patients with therapy-related acute myeloid leukemia with normal karyotype
Blood Cancer Journal (2020)
-
Genetic Pathway in the Pathogenesis of Therapy-Related Myeloid Neoplasms: A Literature Review
Oncology and Therapy (2020)
-
Immunohistochemistry for p53 is a useful tool to identify cases of acute myeloid leukemia with myelodysplasia-related changes that are TP53 mutated, have complex karyotype, and have poor prognosis
Modern Pathology (2017)
