
Abstract
The ossifying renal tumor of infancy is a rare neoplasm diagnosed in the first 2 years of life, predominantly in boys. The neoplasm is primarily characterized by the presence of a large ossifying component. Its most common mode of presentation is hematuria, and it has a uniformly benign behavior. The karyotypic makeup of the process has not been reported. Thus, a study was undertaken and it allowed demonstration of clonal trisomy 4, which was confirmed by the fluorescent in-situ hybridization-probing of two additional archival formalin-fixed, paraffin-imbedded similar tumors. On the basis of the findings in these three cases, it seems that clonal trisomy 4 may be considered as a characteristic of the tumor, which makes it distinct from any other infantile renal tumor.
Similar content being viewed by others

Main
The ossifying renal tumor of infancy is a rare neoplasm originally described by Chatten et al.1 It is diagnosed in the first 2 years of life, predominantly in boys. The neoplasm is primarily characterized by the presence of a large ossifying component.2 Its most common mode of presentation is hematuria, as it usually mushrooms and bulges into the pelvi–caliceal system. This neoplasm has a uniformly benign behavior. One patient with this tumor was followed up for more than 23 years without recurrence.3 The tumor has not been shown to be related to any of the well-established syndromes or infantile renal tumors such as the Wilms tumor or the Mesoblastic Nephroma, albeit the kidney of one case was reported as also harboring intralobar nephroblastomatosis.2 The karyotypic makeup of the process has not been described, which is prompting this communication of our finding of clonal trisomy 4 in cultured tumor cells. This aneuploidy was also detected and confirmed by the fluorescent in-situ hybridization (FISH) probing of the tumor as well as two additional archival formalin-fixed, paraffin-imbedded (FFPE) ossifying renal tumors of infancy.
Case Report
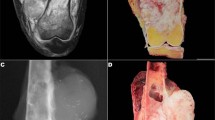
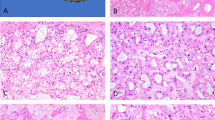
The index case, case A, was that of a 1-year-old male who presented with hematuria and was found to have a 3.5-cm, in maximum diameter, stony hard mass in the upper pole of the right kidney, bulging within the pelvi–caliceal system (Figure 1). The patient underwent a nephrectomy. Histologically, the tumor was heterogeneous, composed mainly of osteoid trabeculae and mature bone lined by osteoblasts and some osteoclasts with no atypical features. Wedged between the osteoid and ossified trabeculae were islands of eosinophilic epithelioid cells. Other spaces contained loose to mildly compact spindle cells (Figure 2). Necrosis and mitotic figures were not seen. The tumor was well demarcated from the surrounding renal tissue that showed compression with tubular atrophy, but no encapsulation. There was no evidence of intra or perilobar nephroblastomatosis. Immunohistochemistry was non-contributory and, in particular, did not detect a nuclear WT1 antigen.

Macroscopic illustration of the nephrectomy specimen in which the ossified tumor is seen bulging into the pelvi–calyceal space (arrow).

Histological illustration of the various components of the tumor. (a) Low-power sample showing the widespread ossification. (b) Higher magnification illustrating the spaces between the osteoid and bony spicules in which epithelioid cells and spindle cells can be seen. (c) A space filled with spindle cells. (d) Osteoid-rich area with epithelioid cells wedged between the spicules. (e) Higher magnification of the epithelioid component of the tumor. (f) Detail of the eosinophilic epithelioid cells.
Samples of the tumor were submitted for chromosomal studies, and, once the results became available, slides from FFPE tissue from two archival cases of similar tumors were submitted for verification studies by FISH. The two archival cases originated from 3- and 4-month-old males (cases B and C). They showed a very similar histology, albeit the amount of osteoid and bone they contained was much greater in the index case, possibly because of the age of the patient (12 months vs 3 and 4 months). Those archival cases had been the object of the original description of the tumor,1 and had been also included in a later review article analyzing nine cases.2
Materials and methods
Pathology
FFPE tissue sections were stained with hematoxylin–phloxine–saffron and hematoxylin and eosin for routine histological assessment. Immunohistochemical studies were conducted using an automated immunostainer (Thermo Scientific, Freemont, CA). The following primary antibodies were used at the dilution recommended by the manufacturer: EMA, β-catenin, CD34, CD99, cytokeratin 8-18, pancytokeratin, vimentin, WT1. All antibodies were obtained from Cell Marque (Rocklin, CA).
Conventional Cytogenetic Analysis
Tumor tissue from the index case was disaggregated manually and enzymatically, then cultured in T-25 flask (Corning, Corning, NY) in conditioned RPMI 1640 medium supplemented with 25% fetal bovine serum for 3 weeks. The medium was changed every 2 days. Metaphase chromosomes for Giemsa banding pattern by trypsin digestion with Wright stain (GTW banding) were prepared according to the standard procedures. Twenty metaphases were karyotyped with CytoVison system (Applied Imaging, Santa Clara, CA), and karyograms were described according to the International System for Human Cytogenetic Nomenclature 2009.4
Interphase FISH Assay
FISH analysis on monolayer interphase nuclei harvested from tissue cultures was undertaken using CEP 4 SpectrumOrange α-satellite DNA probe at the 4p11q11 centromere region of chromosome 4 (Vysis, Abbott Park, IL). A standard FISH protocol was followed. FISH analysis on FFPE tumor specimens was performed on 3-μm tissue sections. The areas of the tumor on the slides were identified and marked by a pathologist. The slides were then de-paraffinized using the Paraffin Pretreatment Reagent Kit III, and hybridized with the probe, overnight, according to the manufacturer’s protocol (Vysis, Abbott Park, IL). The next day, the slides were washed twice in 2 × standard saline citrate (SSC) (Sigma-Aldrich, St Louis, MO) for 2 min at 72°C, followed by 2 × SSC for 2 min at room temperature, and then stained with 4′,6-diamidino-2-phenylindole. Hybridization signals were captured and analyzed with a CytoVison system (Applied Imaging, Santa Clara, CA). Only non-overlapping nuclei with well-defined outline were selected for scoring the hybridization signals. Small, pale, irregular and ambiguous fluorescent deposits in the nuclear area were disregarded. The positive hybridization signals using centromere-specific probes should be well defined with consistence in size and intensity even across different specimens. Ten Wilms tumor and three mesoblastic nephroma specimens were studied as controls. A positive trisomy was accepted if the number of nuclei with three hybridization signals for the centromere probe were greater than 2 s.d.’s from the highest number of nuclei with trisomy present on a slide from the control specimens.5, 6, 7
Results
Immunohistochemistry
No reactivity was detected for EMA, cytokeratin 8-18 and pancytokeratin, vimentin, CD99, or CD34. Anti-WT1 and β-catenin showed only cytoplasmic staining, but no nuclear reactivity was present.
Cytogenetics and FISH Studies on Cultured Tumor Cells from the Index Case
GTW-banding analysis of case A, performed on un-stimulated tumor cells growing in culture for 3 weeks, demonstrated an extra copy of chromosome 4 as the sole clonal abnormality detected in 3 of 20 metaphases analyzed (Figure 3a). Additionally, random loss of chromosomes 6, 8, 13, 15, 16, 17, 18, 19, 20, 21, 22, and Y, gain of chromosome 3, and a questionable structural rearrangement involving the short arm of chromosome 16 were also seen in different cells (data not shown). As those random alterations appeared only a single time, they did not meet the criteria for ‘clonal’ change,4 and were not described in the nomenclature, but their presence was noted for eventual future studies.

Conventional cytogenetic and fluorescent in-situ hybridization (FISH) studies with chromosome 4 centromere probe on the index and archival ossifying renal tumors of infancy. (a) Representative karyotype of index case (case A) shows trisomy 4 as the sole clonal aberration by chromosome analysis (solid arrow), random losses of chromosome 8 and a questionable structural rearrangement involving chromosome 16 (open arrows). (b) Hybridization signals indicative of trisomy 4 demonstrated on nuclei from cultured index case (case A) cells by interphase FISH. (c) Hybridization signals indicative of tetrasomy 4 demonstrated on nuclei from cultured index case (case A) cells by interphase FISH. (d) Hybridization signals indicative of trisomy 4 demonstrated in cells on the formalin-fixed, paraffin-imbedded section from the archival tumor case B.
FISH with CEP4 probe on cultured interphase nuclei, as a different cytogenetic method, was conducted to evaluate chromosome 4 copy number changes in the index tumor. Two abnormal hybridization patterns were observed. In the first pattern, three hybridization signals for the CEP 4 probe indicative of trisomy 4 were seen in three of five hundred nuclei (0.6%) (Figure 3b). In the second pattern, four signals for the CEP4 probe indicative of tetrasomy 4 were present in 7 of 500 nuclei (1.4%) (Figure 3c). This tetrasomy 4 was not detected by conventional chromosome analysis of the tumor. In total, 2% of the analyzed interphase nuclei from cultured tumor cells displayed aneuploidy involving chromosome 4.
FISH Studies on FFPE Slides from Two Archival Tumors
This study was aimed at verifying whether trisomy 4 demonstrated in the index case (case A) represented a cytogenetic event consistently observed in other ossifying renal tumors of infancy. FISH was performed with CEP4 probe on the FFPE slides obtained from the two archival cases baring the same diagnosis (cases B and C). In case B, trisomy 4 was seen in 25 of 200 interphase nuclei (12.5%) analyzed on the FFPE slide (Figure 3d). In case C, the cell morphology was very poor and the conventional criteria for a complete FISH analysis requiring examination of at least 100 nuclei could not be met; only 25 nuclei were available for study, and only 1 nucleus contained signals consistent with trisomy 4. Upon repeating the FISH assays on both archival cases, similar findings were obtained in case B, whereas case C still showed poor cell morphology with only 12 nuclei available and 1 nucleus containing trisomy 4. Tetrasomy 4 seen in the index case was not evident in either of the archival tumors.
Ten Wilms tumor and three mesoblastic nephroma specimens (Supplementary Table 1) were randomly selected from our archive as controls, and FISH with the CEP4 probe was performed on FFPE sections from those cases. A total of 100–200 morphologically acceptable nuclei were analyzed from each of the control specimens. In total, we have evaluated 2350 nuclei on the control FFPE slides. The greatest number of positive trisomy 4 nuclei in the control renal tumors was 1.5% (ranging 0–1.5%); the s.d. was 0.43%; therefore, the cutoff point for trisomy 4 was set at 2.4%, and the cutoff for tetrasomy 4 was set at 0.8%. (Supplementary Table 1 and Supplementary Figure 1). Karyotypes obtained from conventional cytogenetic analysis of those tumors were reviewed after all FISH studies had completed. Neither numerical nor structural aberrations involving chromosome 4 were observed in the randomly selected control specimens. Therefore, we consider monosomy 4, trisomy 4, and tetrasomy 4 when found in the control Wilms tumors and mesoblastic nephromas as non-specific. Using 2.4% as the cutoff, case B (12.5%) is obviously positive for trisomy 4.
Discussion
We report the finding of trisomy 4 identified by both conventional karyotyping and FISH studies in the ossifying renal tumor of infancy. In the index case, we observed trisomy 4 as a clonal aberration in 3 of 20 dividing metaphases cells, but only in 0.6% of non-dividing interphase nuclei. In addition, tetrasomy 4 was only seen in 1.4% of non-dividing nuclei and not detected in the dividing metaphases, which could be due to different growth rate of subgroups of cells. We also observed multiple random chromosomal losses, gains and suspicious structural aberration, but those did not meet the clonality definition and could represent culture artifact or emerging clonal cell populations.4 To confirm that trisomy 4 as a characteristic cytogenetic finding in ossifying renal tumors of infancy instead of being a non-specific feature that can also be observed in other renal neoplasms, we performed the same FISH studies on FFPE sections from randomly selected ten Wilms tumors and three mesoblastic nephromas. All 13 control tumors showed normal disomy for chromosome 4, regardless of the karyotypic findings (Supplementary Table 1 and Supplementary Figure 1). A cutoff value of 2.4% was subsequently calculated.
FISH studies on interphase nuclei, either from cultured cells or on FFPE slides from the three ossifying renal tumors of infancy, revealed trisomy 4 in 0.6% (case A), 11.5% (case B) and 4% (case C) of the nuclei analyzed. Possible artifact hybridization signals in cases A and C cannot be ruled-out due to the relatively low percentage of positivity. However, in the index case, conventional chromosome analysis performed on metaphase cells also demonstrated trisomy 4 in 3 of 20 metaphases (15%), a similar positive finding detected by a different cytogenetic approach, thus providing a confident validation for this case. FISH assay on FFPE slides from case C was repeated several times, but a complete FISH assay with at least 100 nuclei analyzed could not be achieved in any study due to poor cell morphology and ambiguous hybridization signals. The percentages of positive nuclei in these incomplete FISH studies were found to fall outside the cutoff value of 2.4%.
The relative low percentage of the trisomy 4 in the tumor could be attributed to several reasons: First, trisomy 4 might be the driving power of tumorigenesis in the ossifying renal tumor. Only a small group of cells containing trisomy 4 are able to initiate the tumor development and promote the growth, while the majority of the cells have lost the extra copy of chromosome 4 to maintain a non-malignant, less aggressive clinical condition. As a matter of fact, most of the currently available cytogenetic findings were obtained from highly advanced tumors, and very few were identified from benign solid tumors.8 For example, high percentage trisomy 7 and trisomy 17 were seen in papillary renal cell carcinomas. Second, FISH on FFPE slides has critical limitations as opposed to chromosome analysis, such as nuclear overlap and truncation.9, 10, 11 In our study, to circumvent the problem of cellular overlap, we used very thin paraffin section of 3-μm, which has the disadvantage of possibly leading to underestimation of trisomy signals due to nuclear truncation. Among the 13 control cases (Supplementary Table 1), 39 of the 2350 nuclei examined (1.65%) demonstrated non-specific monosomy 4, whereas only 8 nuclei (0.3%) showed non-specific trisomy (8 nuclei, 0.3%) and 1 nucleus (0.04%) showed non-specific tetrasomy.
In addition, it has also been shown in a comparative FISH study between touch preparations and cultured cells that the percentage of aneuploid cells is higher in cultured cells, suggesting that one can be reporting underestimations of aneuploidy when performing interphase nuclei studies on touch preparations and probably also on paraffin sections.12 Hence, accurately and quantitatively assessing chromosome copy number requires laser scanner or confocal microscopy that is usually beyond the means of the average cytogenetics laboratory. Third, previous treatment of the tissue, such as age of the blocks, decalcification and length of formalin fixation may be also implicated. In summary, we are confident to state that in at least two of three ossifying renal tumors of infancy examined (index case and case B), trisomy 4 was consistently observed and seems to be a cytogenetic alteration characteristic of the tumor.
Neoplastic cells showing trisomy 4 as the sole clonal abnormality are very rare. They have been primarily reported in hematological disorders.3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13 Solid tumor cells with this sort of alteration are even more exceptional and only the subject of occasional single, relatively old, case report describing heterogeneous conditions such as thyroid neoplasia,7, 14 uterine leiomyoma,15 ovarian thecoma16 and a parotid tumor metastasizing to the mandible.17 Thus, it is obvious that the presence of trisomy 4 cells as a clonal abnormality, whether isolated or part of more complex karyotypic change, cannot be equated with malignancy or indicative of a cell of origin. This has been shown in the case of other trisomies such as trisomy 7, for example, which can be found in both benign and malignant solid tumors, in inflammatory lesions such as osteoarthritis and rheumatoid arthritis, as well as in apparently normal tissues.18 However, in some contexts, detection of cells trisomic for chromosome 4 in a given neoplastic process may be useful as an additional identifier of the process and could allow better characterization of the lesion, separating it from other congeners, namely, in this particular instance, from Wilms tumor and mesoblastic nephroma.
The biological role trisomy 4 has in the ossifying renal tumors of infancy remains to be determined. Chromosome aberrations have a usually major role in the initiation and progression of benign and malignant tumors.19 Numeric abnormalities of chromosome 4 including both gains and losses have been shown to be associated with tumor stage and lymph node involvement in cases of breast carcinoma.20 Deletion of chromosome 4 or part of chromosome 4 has been reported in esthesioneuroblastoma21 and clear-cell renal cell carcinomas;22 suppression of transformed phenotype and tumorigenicity was seen after the transfer of chromosome 4 into U251 human glioma cells,23 indicating the presence of tumor suppressor genes on chromosome 4.24 On the other hand, amplification or extra copies of chromosome 4, which is the site of wild-type KIT gene, has been commonly observed in many tumors, the significance of which remains unclear.25
In ossifying renal tumor of infancy, whether trisomy 4 is an early event or a late result of increasing genetic instability or other genetic events such as inactivation of tumor suppressor pathways requires further investigation. It is even possible that trisomy 4 not be as significant as the role of the chromosomal instability (CIN) involving gain or loss of multiple whole chromosomes or fractions of multiple chromosomes in the initiation of the neoplastic process. While karyotyping the index case, random non-clonal losses and gains of different chromosomes were observed (data not shown), which could indicate an enhanced frequency of chromosome mis-segregation or increased level of genetic diversity. Moreover, although observed by FISH study alone, tetrasomy 4 in the index case might reflect inability of the cells to faithfully segregate equal chromosome complements to two daughter cells during mitosis. Thus, genetic or cellular events underlying CIN, such as defects in chromosome cohesion, mitotic spindle checkpoints, control of supernumerary centrosomes, kinetochore–microtubule attachment dynamics, and cell-cycle regulation, might be upstream controllers involved in the initiation of this tumor.26
Before closing we would like to add that the immunohistochemical testing we have conducted essentially consistently yielded negative results.
In conclusion, we can confidently state that in at least two of three ossifying renal tumors of infancy we have examined, trisomy 4 was consistently observed and seems to be a cytogenetic alteration characteristic of the tumor. Detection of cells trisomic for chromosome 4 in this neoplastic process may be useful as an additional identifier of the process and could allow better characterization of the lesion, separating it from other congeners, Wilms tumor and mesoblastic nephroma in particular.
References
Chatten J, Cromie WJ, Duckett JW . Ossifying tumor of infantile kidney: report of two cases. Cancer 1980;45:609–612.
Sotelo-Avila C, Beckwith JB, Johnson JE . Ossifying renal tumor of infancy: a clinicopathologic study of nine cases. Pediatr Pathol Lab Med 1995;15:745–762.
Ito J, Shinohara N, Koyanagi T et al Ossifying renal tumor of infancy: the first Japanese case with long-term follow-up. Pathol Int 1998;48:151–159.
Shaffer LG . ISCN 2009: An International System for Human Cytogenetic Nomenclature (2009): Recommendations of the International Standing Committee on Human Cytogenetic Nomenclature 1st edn S. Karger Publishers, Inc. Connecticut, USA, 2009.
Blanco R, Lyda M, Davis B et al Trisomy 3 in gastric lymphomas of extranodal marginal zone B-cell (mucosa-associated lymphoid tissue) origin demonstrated by FISH in intact paraffin tissue sections. Hum Pathol 1999;30:706–711.
Wotherspoon AC, Finn TM, Isaacson PG . Trisomy 3 in low-grade B-cell lymphomas of mucosa-associated lymphoid tissue. Blood 1995;85:2000–2004.
Visakorpi T, Hyytinen E, Kallioniemi A et al Sensitive detection of chromosome copy number aberrations in prostate cancer by fluorescence in situ hybridization. Am J Pathol 1994;145:624–630.
Mitelman F, Heim S . The significance of karyotypic changes in human neoplasia. Haematologica 1987;72 (6 Suppl):29–33.
Lee W, Han K, Harris CP et al Detection of aneuploidy and possible deletion in paraffin-embedded rhabdomyosarcoma cells with FISH. Cancer Genet Cytogenet 1993;68:99–103.
Afify A, Bland KI, Mark HF . Fluorescent in situ hybridization assessment of chromosome 8 copy number in breast cancer. Breast Cancer Res Treat 1996;38:201–208.
D'Alessandro I, Zitzelsberger H, Hutzler P et al Numerical aberrations of chromosome 7 detected in 15 microns paraffin-embedded tissue sections of primary cutaneous melanomas by fluorescence in situ hybridization and confocal laser scanning microscopy. J Cutan Pathol 1997;24:70–75.
Nadal M, Pera G, Gomez-Zaera M et al Tissue imprints or primary cultures: which strategy to use to study cytogenetic clonality? Diagn Mol Pathol 2005;14:243–246.
Berger R, Nguyen-Khac F . [Chromosomal abnormalities and Waldenstrom macroglobulinemia]. Pathol Biol (Paris) 2008;56:400–406.
Bondeson L, Bengtsson A, Bondeson AG et al Chromosome studies in thyroid neoplasia. Cancer 1989;64:680–685.
Nilbert M, Heim S, Mandahl N et al Trisomy 12 in uterine leiomyomas. A new cytogenetic subgroup. Cancer Genet Cytogenet 1990;45:63–66.
Mrozek K, Limon J, Debniak J et al Trisomy 12 and 4 in a thecoma of the ovary. Gynecol Oncol 1992;45:66–68.
Manor E, Sion-Vardy N, Bodner L . Cytogenetic and fluorescence in situ hybridization analysis of a basal cell adenocarcinoma of the mandible. Cancer Genet Cytogenet 2006;166:186–188.
Broberg K, Toksvig-Larsen S, Lindstrand A et al Trisomy 7 accumulates with age in solid tumors and non-neoplastic synovia. Genes Chromosomes Cancer 2001;30:310–315.
Gancberg D, Scourneau M, Verdebout JM et al Detection of extra chromosomes 12 by fluorescent in situ hybridization (FISH) in ovarian stromal tumors. Study of 12 cases and review of the literature. Ann Pathol 2001;21:393–398.
Persons DL, Robinson RA, Hsu PH et al Chromosome-specific aneusomy in carcinoma of the breast. Clin Cancer Res 1996;2:883–888.
Riazimand SH, Brieger J, Jacob R et al Analysis of cytogenetic aberrations in esthesioneuroblastomas by comparative genomic hybridization. Cancer Genet Cytogenet 2002;136:53–57.
Reutzel D, Mende M, Naumann S et al Genomic imbalances in 61 renal cancers from the proximal tubulus detected by comparative genomic hybridization. Cytogenet Cell Genet 2001;93:221–227.
Pershouse MA, Ligon AH, Pereira-Smith OM et al Suppression of transformed phenotype and tumorigenicity after transfer of chromosome 4 into U251 human glioma cells. Genes Chromosomes Cancer 1997;20:260–267.
Arribas R, Risques RA, Gonzalez-Garcia I et al Tracking recurrent quantitative genomic alterations in colorectal cancer: allelic losses in chromosome 4 correlate with tumor aggressiveness. Lab Invest 1999;79:111–122.
Sihto H, Sarlomo-Rikala M, Tynninen O et al KIT and platelet-derived growth factor receptor alpha tyrosine kinase gene mutations and KIT amplifications in human solid tumors. J Clin Oncol 2005;23:49–57.
Chan JY . A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 2011;7:1122–1144.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Supplementary information
Rights and permissions
About this article
Cite this article
Liu, J., Guzman, M., Pawel, B. et al. Clonal trisomy 4 cells detected in the ossifying renal tumor of infancy: study of 3 cases. Mod Pathol 26, 275–281 (2013). https://doi.org/10.1038/modpathol.2012.120
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2012.120

{kind=link}