
Abstract
The recent development of inhibitors of key immune response proteins has revolutionized the therapy of autoimmune diseases; these immunomodulator agents include monoclonal antibodies and receptor antagonists. However, as with all therapies, these new agents are not without side effects and complications. In particular, anti-tumor necrosis factor alpha (TNFα) agents have been reported to be associated with an increased incidence of lymphoproliferative disorders, infections, and vasculitis. We evaluated the clinicopathological features of 18 cases of immunomodulator agent-related lymphoproliferative disorders (IAR-LPD) from several institutions. These included 6 cases of B-cell lymphoma, 2 cases of T-cell lymphoma, 3 cases of classical Hodgkin lymphoma, and 7 atypical lymphoid proliferations that did not fulfill diagnostic criteria for lymphoma; two of the latter regressed after discontinuation of the immunomodulator agent therapy. All eight lymphoma patients with available information had also received prior chemotherapy (methotrexate or 6-mercaptopurine). EBV was strongly associated with the B-cell and classical Hodgkin lymphomas. This case series illustrates that a broad range of lymphoid proliferations can occur after immunomodulator agent therapy and that these immunomodulator agent-related lymphoproliferative disorders have considerable overlap with other well-defined lymphoproliferative diseases associated with iatrogenic immunosuppression. Further study is warranted to evaluate how these therapies interact with other immunosuppressive agents and the underlying abnormal immune system to enhance the development of lymphomas and atypical lymphoid proliferations.
Similar content being viewed by others

Main
The treatment of autoimmune diseases has undergone a revolution in recent years. Numerous new therapies have been developed based on inhibition of pro-inflammatory cytokines, particularly tumor necrosis factor alpha (TNFα). These immunomodulator agents are mostly humanized monoclonal antibodies that block receptor engagement and subsequent downstream activation; fusion proteins and receptor antagonists have also been developed that block receptor activation. Immunomodulator agents are effective against a number of autoimmune diseases and can also serve as inhibitors of organ rejection (Table 1). Blockage of the TNFα pathway leads to a number of anti-inflammatory effects, including reduction of other pro-inflammatory cytokines, and modulation of the stromal and vascular microenvironment. The ultimate clinical goal of these therapies is to decrease the inflammatory response and to suppress autoimmune damage to target tissues, such as the synovium, the cartilage, the skin, and the gastrointestinal epithelium.1 Blockage of CD25 (interleukin-2 receptor alpha chain) prevents the expansion of autoreactive T-cells, whereas inhibition of CD11a (a subunit of lymphocyte function-associated antigen-1 (LFA-1)) suppresses the activation of autoreactive T-cells. Competitive inhibition of interleukin 1 (IL-1) binding to its receptor can inhibit inflammatory responses in the synovium.2, 3 However, as with all therapies, these new therapies are not without side effects and complications. TNFα inhibitors,4, 5, 6, 7 as well as other monoclonal antibody-based anti-inflammatory agents,8, 9, 10, 11 have been associated with the development of lymphoproliferative disorders. Most of these reports have involved small numbers of patients. In some cases, the lymphoproliferative diseases regressed upon withdrawal of the therapy, implicating immunomodulator agents in their pathogenesis.4 However, most large epidemiological studies have failed to show an increased risk of lymphomas in patients treated with immunomodulator agents.5, 6, 7 These analyses are partly confounded by the fact that the incidence of lymphomas is increased in rheumatoid arthritis (between two- and fourfold over the general population)8 and the concomitant administration of other drugs known to be associated with lymphoproliferative diseases, such as methotrexate.9 Nevertheless, TNFα antagonists are included as causative agents in the group of lymphoproliferative disorders associated with iatrogenic immunosuppression in the 2008 WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues.10 The purpose of this study is to evaluate the clinicopathological features of 18 cases of lymphoproliferative disorders arising in patients treated with TNFα, CD11a, CD25, and/or IL-1 receptor antagonists (immunomodulator agent-related lymphoproliferative disorders (IAR-LPD)).
Materials and methods
Cases of atypical or neoplastic lymphoid proliferations in patients after documented treatment with any of the drugs in Table 1 were obtained from multiple institutions. In all cases, clinical information and pathological material was reviewed with respect to institutional safeguards for patient privacy. Demographic information, medication information (type and duration) and available follow-up information were recorded. Histological sections were reviewed by two of the authors (RPH and DOM), including hematoxylin and eosin (H&E) stains on all cases and results of immunohistochemical staining when available. Cases were categorized as lymphomas or as lymphoid proliferations not diagnostic of lymphoma; given the diverse nature of the latter, the features of these cases are described individually in the Results section below. The antibodies evaluated on a case-by-case basis included: CD2, CD3, CD4, CD5, CD7, CD8, βF1, CD20, CD30, PAX5, bcl-6, bcl-2, CD10, MUM-1, ALK-1, EMA, TIA-1, clusterin, and cutaneous lymphocyte antigen (CLA). In situ hybridization for EBV encoded RNA (EBER) was carried out in 16 cases, where material was available.
Gene Rearrangement Studies
In all cases where blocks were available, gene rearrangement studies were carried out at Clairent, Aliso Viejo, CA, USA. Briefly, formalin-fixed paraffin embedded tissue sections were deparaffinized with xylene and DNA extracted using QIAamp DNA Mini kit (Qiagen, Valencia, CA, USA). Amplification of IGH, TCR-β, and TCR-γ was carried out using fluorogenically-labeled primer sets specific for junctional (J) region (and diversity (D) in TCR-β PCR) and framework determinants of the variable (V) region of the IGH, TCR-β, and TCR-γ genes according to published protocols.11 PCR products were separated by capillary gel electrophoresis and the size, number, and fluorescent intensity of the PCR product peaks generated were used to determine the presence of a clonal gene rearrangement according to previously described criteria for clonality.12 In some cases, gene rearrangement studies had been carried out at individual institutions according to the institutional protocol.
Results
The age of patients ranged from 24 to 75 years (median 55 years). There were 10 males and 8 females. The cases submitted encompassed a broad range of atypical reactive and overtly malignant lymphoid proliferations. The patients had a variety of autoimmune disorders, highlighting the broad use of these agents. The most common diseases treated were rheumatoid arthritis (5 patients), Crohn's disease (4 patients), and psoriasis (4 patients). Other diseases in this series were ulcerative colitis (2 cases), multiple sclerosis (1 case), and ankylosing spondylitis (1 case and an additional case with concurrent rheumatoid arthritis). One patient had pyogenic arthritis, pyoderma gangrenosum, and acne syndrome (PAPA), an inherited autosomal dominant disorder associated with mutation in the CD2 binding protein 1 gene (CD2BP1) on chromosome 15.13 The IAR-LPD were diagnosed a median of 2 years following institution of the immunomodulator agent therapy (range 6 weeks to 7 years among the nine patients with available data). All seven patients with atypical lymphoid proliferations were male, whereas most of the lymphomas (8/11, 72%) occurred in female patients (P=0.004). The lymphoma cases also included many (5/11, 45%) rheumatoid arthritis patients, whereas none of the seven atypical lymphoid proliferation cases occurred in patients with rheumatoid arthritis; the latter finding did not reach statistical significance (P=0.10). In all, 9/11 (82%) lymphoma patients and 4/7 (57%) atypical lymphoid proliferation patients were treated with anti-TNFα therapies. A total of 6/8 (75%) lymphoma patients with available information had been treated with methotrexate in addition to the immunomodulator agents; the other 2/8 patients had received 6-mercaptopurine, with or without steroids. In contrast, 0/5 atypical lymphoid proliferation patients with available information had received methotrexate (P=0.02 compared with lymphoma patients); 3/5 patients had received other therapies (steroids in 2/5 patients and interferon in 1/5 patient).
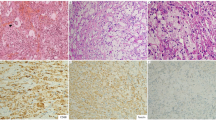
Of the 18 IAR-LPD cases evaluated in this series, 11 were diagnosed as lymphoma (3 diffuse large B-cell lymphoma, including 1 that arose 3 years following a diagnosis of lymphoplasmacytic lymphoma; 3 classical Hodgkin lymphoma; 2 T-cell lymphomas; 2 follicular lymphomas; and 1 extranodal marginal zone B-cell lymphoma). Unusual features of these lymphomas included EBV positivity of one follicular lymphoma (Case 4), ALK positivity of a CD2+, CD3+ cutaneous anaplastic large cell lymphoma (Case 10), and presentation of classical Hodgkin lymphoma as an intestinal mass (in Case 11 as the initial presentation and in Case 17 as a second malignancy occurring 4 years after follicular lymphoma). The subcutaneous panniculitis-like T-cell lymphoma (Case 2) was unusual in that it expressed CD4 rather than CD8. This case was CD56 negative, making the possibility of a cutaneous γ–δ T-cell lymphoma unlikely, and did not involve the dermis, excluding a cutaneous small/medium pleomorphic T-cell lymphoma. The clinical and pathological features of the lymphoma cases are summarized in Table 2 and representative images from examples of lymphoma cases are shown in Figure 1. One lymphoma case (Case 5) was previously reported.14

Examples of immunomodulator agent-related lymphoproliferative disorder (IAR-LPD) lymphoma cases. (a) Case 4, follicular lymphoma. The lymph node is effaced by neoplastic follicles typical of follicular lymphoma (hematoxylin and eosin (H&E)). (b) Case 2, subcutaneous panniculitis-like T-cell lymphoma. The atypical small to medium-sized lymphoid cells infiltrate subcutaneous adipose tissue, with characteristic ‘rimming’ of adipocytes (H&E). (c, d): Case 7, diffuse large B-cell lymphoma, showing sheets of large cells (H&E, c) that are positive for EBER (d). (e, f): Case 15, classical Hodgkin lymphoma, with neoplastic Reed–Sternberg cells in a mixed inflammatory background (H&E, e). The Reed–Sternberg cells are positive for CD30 (f).
The other seven cases were characterized as atypical lymphoid proliferations that raised the possibility of or (Cases 3 and 18) were suspicious for lymphoma, but were not absolutely diagnostic of lymphoma, based on small sample size, results of molecular studies, and/or review by two authors (RPH and DOM). The clinical and pathological features of these non-lymphoma cases are summarized in Table 3 and images from two example cases are shown in Figure 2. Detailed histological descriptions of the seven non-lymphoma cases are provided below.

Examples of immunomodulator agent-related lymphoproliferative disorders (IAR-LPD) that are atypical lymphoid proliferations not diagnostic of lymphoma. (a, b): Case 1, an atypical nodal follicular and paracortical hyperplasia that distorts the lymph node architecture (hematoxylin and eosin (H&E), a). CD20 (b) is positive in follicles, as well as in many cells between the follicles. (c, d): Case 14, a polyclonal EBV+ lymphoproliferative disorder that regressed on withdrawal of efalizumab therapy. There is effacement by a polymorphous lymphoid proliferation with many large, atypical cells (H&E,c). EBV encoded RNA (EBER) is positive in numerous medium and large cells (d).
Case 1: The lymph node architecture was distorted by a marked proliferation of reactive-appearing lymphoid follicles, as well as a vaguely nodular paracortical hyperplasia with a polymorphous cell population and a moderate vascular proliferation (Figure 2a and b). Staining for EBER was negative. The patient's diffuse lymphadenopathy completely regressed on reduction in dosage of the immunomodulatory drug (daclizumab) and has not recurred at the latest follow up of 2 years.
Case 3: The axillary lymph node contained a proliferation of atypical cells, predominantly comprising CD3+, CD5+, CD4+, and βF1+ T-cells with downregulation of CD2 and CD7 by immunohistochemistry; some medium to large cells expressed CD30. EBER was negative and IgH and TCR gene rearrangement studies by PCR were negative. Serological studies showed that the patient was HIV negative and was hepatitis A, B, and C negative. EBV IgG and IgM serologies were also negative. Follow up is not available on this patient.
Case 8: The needle biopsy of lymph node showed a proliferation of small, atypical lymphoid cells with areas of geographical necrosis. The atypical cells were CD3+ T-cells that were CD2+, CD45RO+, and CD7+, with rare CD30+, ALK1−, and CD15− large cells. About 40% of the T-cells were CD8+; CD4 staining was suboptimal and could not be interpreted. EBV studies are not available on this patient. Although the findings were not felt to be diagnostic of lymphoma, the patient was treated empirically for lymphoma with chemotherapy; further follow up is not available.
Case 9: The skin contained an abnormal lymphohistiocytic infiltrate with vasculitis; small blood vessels were infiltrated by CD3+ and CD2+ T-cells, with a mixture of CD4+ and CD8+ subsets. Small areas of necrosis were present. Some T-cells showed apparent decreased CD5, βF1, and CD7 staining. TIA1 was positive in many of the lymphocytes and CD56 was positive in rare cells. Rare CD30+ large cells were also noted. EBER was negative and molecular studies did not detect a clonal B-cell or T-cell population. Infliximab was discontinued, but the patient developed brain lesions 3 years later with biopsies showing a similar lymphohistiocytic infiltrate. The patient was treated with cyclophosphamide, rituximab, and steroids and is alive with persistent disease 3 years after diagnosis.
Case 14: This patient experienced a 40-pound weight loss, fevers, night sweats, and developed diffuse lymphadenopathy. An inguinal lymph node biopsy was enlarged with multiple areas of geographical necrosis and a surrounding heterogeneous admixture of small and large lymphocytes and histiocytes, including numerous immunoblasts. Most of the small lymphocytes were CD3+, CD5+ T-cells with CD4+ cells outnumbering CD8+ cells, but there were also numerous large CD20+, MUM1+, CD79a+ B-cells that were frequently CD30+ and were EBER+ (Figures 2c and d). CD15 was negative. Biopsy of an axillary lymph node performed a few days later showed similar features. Molecular studies did not detect a clonal B-cell or T-cell population. Blood EBV DNA quantification by PCR was positive at 1441 copies/ml. The overall findings resembled those of an EBV+ lymphoproliferative disease occurring in the setting of immunosuppression. Following discontinuation of the immunomodulatory drug (efalizumab), the patient's lymphadenopathy and other symptoms gradually resolved. At the latest follow up (4 months after presentation), he has undetectable blood EBV DNA and no lymphadenopathy on CT scans.
Case 16: The symptoms of Crohn's disease worsened for this patient 1 year after therapy began with infliximab, prompting endoscopy and biopsy of a nodular-appearing and ulcerated ileum. On histological examination, there were prominent reactive-appearing germinal centers and numerous intraepithelial lymphocytes. Many CD20+ small B-cells were present outside the germinal centers that did not show co-expression of CD5, CD43, or cyclinD1 and had a low Ki67 proliferation index. EBER was negative. Frequent plasma cells were polytypic for κ and λ by in situ hybridization. Molecular studies did not detect a clonal B-cell or T-cell population. The findings were interpreted as an atypical B-cell-rich lymphoid proliferation. As these findings could not be definitively attributed to the infliximab, the drug was not discontinued and the patient remains symptomatic with severe abdominal pain after 2 months.
Case 18: This patient presented with a 4-week history of enlarged cervical and bilateral axillary lymph nodes, as well as a low-grade fever. The excised lymph nodes showed a proliferation of atypical follicles and a prominent interfollicular proliferation of small, irregular B-cells, including frequent B-cells that expressed BCL6, CD10, and weakly expressed BCL2; some follicles also contained a similar population of small, atypical cells and there was faint staining of some follicles for BCL2. Numerous eosinophils were also present in the interfollicular region. Flow cytometry showed a possible small κ-restricted CD20 bright+ B-cell population amidst a polytypic B-cell population, but PCR study failed to reveal an IGH or TCR clonal rearrangement nor could a BCL2 gene rearrangement be detected. EBER was negative and the overall findings were felt to be highly suspicious for a low-grade follicular lymphoma. No follow up is available.
Discussion
These cases illustrate that a broad range of lymphoid proliferations are associated with a relatively new class of agents that block pathways involved in immune stimulation and are used to treat autoimmune or other inflammatory disorders. Although the majority of our cases were associated with anti-TNFα therapy, other biological immune response modifiers with different mechanisms of action (anti-CD25, anti-CD11a, and anti-IL-1 receptor) were also associated with a similar spectrum of pathology. The IAR-LPDs have considerable overlap with other iatrogenic lymphoid proliferations, such as those seen in post-transplant settings and those seen in association with methotrexate therapy. Indeed, the majority (6/8) of the lymphoma patients in our series, vs none of the patients with atypical lymphoid proliferations, had received methotrexate. As with other lymphoid proliferations associated with immunosuppression, EBV was present in many (7/17) of our tested cases, suggesting that EBV may have a permissive or even etiological role in the development of IAR-LPD. In particular, most of the B-cell-derived lymphomas with evaluable material (6/9 cases, including 2/3 diffuse large B-cell lymphomas, 1/2 follicular lymphomas, 0/1 marginal zone lymphoma, and 3/3 classical Hodgkin lymphomas) expressed EBV. To our knowledge, this is the first series of immunomodulator agent associated lymphomas to show high EBV prevalence; previous studies did not report EBV data on most patients.15 In contrast, EBV was not seen in either of the two T-cell lymphomas. Among the non-lymphoma cases, only the case with a prominent B-cell expansion (Case 14) that resembled a polymorphous post-transplant lymphoproliferative disorder was EBV positive.
It is important to note that rheumatoid arthritis itself is associated with an increased risk of lymphoma, the most common type of which is diffuse large B-cell lymphoma. There are conflicting data as to whether Crohn's disease or ulcerative colitis patients bear an increased risk of lymphoma, whereas lymphoma is not known to be increased in psoriasis patients.16 Immunomodulatory therapies are often used to treat aggressive autoimmune diseases that have failed other therapies. Thus, it is possible that these IAR-LPDs arose in patients with severe disease activity that required more aggressive therapy rather than effects of the therapy itself; indeed, the risk of lymphoma appears to be higher in rheumatoid arthritis patients with greater disease activity.17 However, an iatrogenic etiology appears more likely, as the incidence of EBV in rheumatoid arthritis-associated lymphomas is low.18, 19 Methotrexate, used to treat rheumatoid arthritis and other autoimmune diseases, has been closely associated with the development of lymphomas in rheumatoid arthritis patients. These methotrexate-associated lymphomas are most often classical Hodgkin lymphoma and diffuse large B-cell lymphoma that are EBV-positive, similar to the most prevalent lymphomas in our series.9, 20 It is controversial whether immunomodulator agents increase the risk of lymphoma over and above that intrinsic to rheumatoid arthritis treated with other immunosuppressive therapies. In contrast, hepatosplenic γ–δ T-cell lymphoma, has been strongly associated with Crohn's disease patients treated with infliximab.21
Two features argue that immunomodulator agents are associated with the development of lymphoid proliferations: the temporal association of the lymphoid proliferation with the immunomodulator therapy and regression of the proliferation upon withdrawal of the drug. Among the eight patients in our series in whom the time frame of drug administration was known, three patients developed the lymphoproliferative disease 1 year or less after starting the immunomodulator therapy. Other series have also reported a relatively short latency (as short as 8 weeks) between infliximab or etanercept administration and lymphoma development.15 Moreover, in two of our patients (Case 1 and Case 14), non-lymphomatous lymphoid proliferations resolved upon cessation of the immunomodulator therapy. Regression of lymphoid proliferations after cessation of therapy has been reported with several immunomodulator agents, including etanercept,4, 22 as well as infliximab and adalimumab.23 It is uncertain if cessation of immunomodulator agent therapy can cause regression of associated lymphomas; one follicular lymphoma case in our series progressed in spite of cessation of infilximab therapy and a case of aggressive T-cell lymphoma after etanercept therapy failed to respond to cessation of therapy and progressed rapidly.24 However, regression of a diffuse large B-cell lymphoma after cessation of in fliximab without the use of cytotoxic chemotherapy has been reported.15 Thus, it is possible that some of these IAR-LPD may respond to cessation of the immunomodulator therapy, similar to the regression of some methotrexate-associated lymphomas.25 As with post-transplant lymphoproliferative disorders, it is likely that the non-lymphomatous, polyclonal lymphoid proliferations would be more responsive to cessation of immunomodulator therapy than would the monoclonal lymphomas. In our series, the response of two atypical lymphoid proliferation patients and lack of response of one lymphoma patient to immunomodulator agent therapy withdrawal support this hypothesis.
The mechanism by which immunomodulator agents may increase the risk of lymphoma is uncertain. One possibility is that the immunomodulator agents provide an additive risk to other immunosuppressive therapies (concurrent or antecedent). Overexuberant immunosuppression may decrease immune surveillance and allow the expansion of an (often EBV-driven) clonal B-cell population, akin to the development of post-transplant lymphoproliferative disorders. According to such a hypothesis, the immunomodulator agent would represent the ‘straw that broke the camel's back’ in immune suppression that allows the development of a lymphoma or an uncontrolled polyclonal or oligoclonal lymphoid proliferation. Alternatively, immunomodulator agents may cause lymphoma by their specific action on the immune system. One recent study has shown that TNFα blockade reduces the ability of lymphoid tissue to form germinal centers and decreases the number of follicular dendritic cells, raising the possibility that a perturbed germinal center environment induced by TNFα blockade may promote the emergence of a neoplastic B-cell clone.26 Specific mechanisms by which blockade of CD25, CD11a, or IL-1 receptor signaling may promote lymphoma are unknown. In our series, anti-CD11a therapy was associated with lymphomas (two cases) and non-lymphomatous lymphoid proliferations (one case), with all cases positive for EBV. Further study is warranted to determine if particular types of IAR-LPD are associated with specific immunomodulator agent therapies, exposure to other immunosuppressive treatments, or the particular underlying disease. Of note, EBV was strongly associated with the lymphomas in our series (6/11 cases), whereas it was present in only 1/6 atypical lymphoid proliferations; this finding suggests that EBV may not have an important role in the pathogenesis of the polyclonal atypical lymphoid proliferations that occur in patients treated with immunomodulator agnents.
In conclusion, the main focus of this study is to describe the diversity of lymphoid proliferations and lymphomas that can occur in patients treated with immunomodulator agents. These proliferations encompass B-cell lymphomas, classical Hodgkin lymphoma, T-cell lymphomas, and unusual atypical lymphoid proliferations. Drug withdrawal can, in at least some cases, be sufficient to cause abnormal lymphoid proliferations to regress. It is likely that as additional immunomodulator agents are developed and as more patients are treated with this class of drugs, the frequency of IAR-LPD will increase. Further study is warranted to elucidate the mechanisms by which these immunomodulator agents may promote the development of lymphoproliferative diseases. It also remains to be determined what roles are played by the intrinsically abnormal immune system in these treated patients and other antecedent or concurrent immunosuppressive agents.
References
Pucino Jr F, Harbus PT, Goldbach-Mansky R . Use of biologics in rheumatoid arthritis: where are we going? Am J Health Syst Pharm 2006;63:S19–S41.
Bresnihan B, Alvaro-Gracia JM, Cobby M, et al. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum 1998;41:2196–2204.
Van Lent PL, Van De Loo FA, Holthuysen AE, et al. Major role for interleukin 1 but not for tumor necrosis factor in early cartilage damage in immune complex arthritis in mice. J Rheumatol 1995;22:2250–2258.
Park SH, Kim CG, Kim JY, et al. Spontaneous regression of EBV-associated diffuse lymphoproliferative disease in a patient with rheumatoid arthritis after discontinuation of etanercept treatment. Rheumatol Int 2008;28:475–477.
Leonardi CL, Toth D, Cather JC, et al. A review of malignancies observed during efalizumab (Raptiva) clinical trials for plaque psoriasis. J Dermatol 2006;213:204–214.
Askling J, Baecklund E, Granath F, et al. Anti-TNF therapy in RA and risk of malignant lymphomas Relative risks and time-trends in the Swedish Biologics Register. Ann Rheum Dis 2009;68:648–653.
Leombruno JP, Einarson TR, Keystone EC . The safety of anti-tumor necrosis factor treatments in rheumatoid arthritis: meta and exposure adjusted pooled analyses of serious adverse events. Ann Rheum Dis 2009;68:1136–1145.
Thomas E, Brewster DH, Black RJ, et al. Risk of malignancy among patients with rheumatic conditions. Int J Cancer 2000;88:497–502.
Kamel OW, van de Rijn M, Weiss LM, et al. Brief report: reversible lymphomas associated with Epstein-Barr virus occurring during methotrexate therapy for rheumatoid arthritis and dermatomyositis. N Engl J Med 1993;328:1317–1321.
Gaulard P SS, Harris NL, Jaffe ES, et al. Other iatrogenic immunodeficiency-associated lymphoproliferative disordersd. In: Swerdlow SH CE, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW (ed). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues Vol., 4th edn. IARC: Lyon, 2008;350–351.
van Dongen JJ, Langerak AW, Bruggemann M, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003;17:2257–2317.
Sprouse JT, Werling R, Hanke D, et al. T-cell clonality determination using polymerase chain reaction (PCR) amplification of the T-cell receptor gamma-chain gene and capillary electrophoresis of fluorescently labeled PCR products. Am J Clin Pathol 2000;113:838–850.
Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet 2002;11:961–969.
Podolsky DK, Gonzalez G, Hasserjian RP . Case 8-2006: A 71-year-old woman with Crohn's disease and altered mental status. N Engl J Med 2006;354:1178–1184.
Brown SL, Greene MH, Gershon SK, et al. Tumor necrosis factor antagonist therapy and lymphoma development: twenty-six cases reported to the Food and Drug Administration. Arthritis Rheum 2002;46:3151–3158.
Askling J, Brandt L, Lapidus A, et al. Risk of haematopoietic cancer in patients with inflammatory bowel disease. Gut 2005;54:617–622.
Baecklund E, Ekbom A, Sparen P, et al. Disease activity and risk of lymphoma in patients with rheumatoid arthritis: nested case-control study. BMJ 1998;317:180–181.
Baecklund E, Sundstrom C, Ekbom A, et al. Lymphoma subtypes in patients with rheumatoid arthritis: increased proportion of diffuse large B cell lymphoma. Arthritis Rheum 2003;48:1543–1550.
Kamel OW, Holly EA, van de Rijn M, et al. A population based, case control study of non-Hodgkin's lymphoma in patients with rheumatoid arthritis. J Rheumatol 1999;26:1676–1680.
Kamel OW, Weiss LM, van de Rijn M, et al. Hodgkin's disease and lymphoproliferations resembling Hodgkin's disease in patients receiving long-term low-dose methotrexate therapy. Am J Surg Pathol 1996;20:1279–1287.
Rosh JR, Gross T, Mamula P, et al. Hepatosplenic T-cell lymphoma in adolescents and young adults with Crohn's disease: a cautionary tale? Inflamm Bowel Dis 2007;13:1024–1030.
Hurley MY, George MN, Leonardi CL, et al. A transient benign lymph node-based proliferation of T-cells simulating non-Hodgkin lymphoma in a patient with psoriasis treated with tumor necrosis factor alpha and CD11a antagonists. Diagn Pathol 2008;3:13.
Deng A, Harvey V, Sina B, et al. Interstitial granulomatous dermatitis associated with the use of tumor necrosis factor alpha inhibitors. Arch Derm 2006;142:198–202.
Adams AE, Zwicker J, Curiel C, et al. Aggressive cutaneous T-cell lymphomas after TNF-alpha blockade. J Am Acad Dermatol 2004;51:660–662.
Moseley AC, Lindsley HB, Skikne BS, et al. Reversible methotrexate associated lymphoproliferative disease evolving into Hodgkin's disease. J Rheumtol 2000;27:810–813.
Anolik JH, Ravikumar R, Barnard J, et al. Cutting edge: anti-tumor necrosis factor therapy in rheumatoid arthritis inhibits memory B lymphocytes via effects on lymphoid germinal centers and follicular dendritic cell networks. J Immunol 2008;180:688–692.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Hasserjian, R., Chen, S., Perkins, S. et al. Immunomodulator agent-related lymphoproliferative disorders. Mod Pathol 22, 1532–1540 (2009). https://doi.org/10.1038/modpathol.2009.131
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2009.131
Keywords
This article is cited by
-
Small intestinal follicular lymphoma induced by methotrexate: a case report
BMC Gastroenterology (2021)
-
Epstein–Barr virus peptides derived from latent cycle proteins alter NKG2A + NK cell effector function
Scientific Reports (2020)
-
Lymphopenia at diagnosis predicts survival of patients with immunodeficiency-associated lymphoproliferative disorders
Annals of Hematology (2020)
-
Methotrexate-associated lymphoproliferative disorder in the stomach and duodenum: a case report
BMC Gastroenterology (2019)
-
Other immunomodulatory agent-related lymphoproliferative diseases: a single-center series of 72 biopsy-confirmed cases
Modern Pathology (2018)
