
Abstract
Desmoplasia is a fibro-inflammatory process and a well-established feature of pancreatic cancer. A key contributor to pancreatic cancer desmoplasia is the pancreatic stellate cell. Various in vitro and in vivo methods have emerged for the isolation, characterization, and use of pancreatic stellate cells in models of cancer-associated fibrosis. In addition to cell culture models, genetically engineered animal models have been established that spontaneously develop pancreatic cancer with desmoplasia. These animal models are currently being used for the study of pancreatic cancer pathogenesis and for evaluating therapeutics against pancreatic cancer. Here, we review various in vitro and in vivo models that are being used or have the potential to be used to study desmoplasia in pancreatic cancer.
Similar content being viewed by others


Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal malignancies, with a median survival of ~6 months and a 5-year survival rate of <8%.1 Worldwide, PDAC causes more than 331 000 deaths per year, making it the seventh leading cause of cancer-related deaths.2 Many factors contribute to the poor prognosis of PDAC, including both intrinsic and acquired chemoresistance and extensive desmoplasia. The fibro-inflammatory or desmoplastic reaction in PDAC is unique among solid tumors. Broadly, desmoplasia consists of cellular and non-cellular components. The cellular components include cancer-associated pancreatic stellate cells (PSCs) or myofibroblast-like cells and immune cells.3, 4 In the normal pancreas, stellate cells are in a quiescent state (non-activated) and are characterized by the presence of vitamin A-containing lipid droplets in their cytoplasm. Upon pancreatic inflammation and injury, quiescent PSCs become activated, lose vitamin A-containing lipid droplets and undergo various molecular and functional changes, which are reviewed elsewhere.5 A study by Apte et al showed that PDAC stroma consists of activated PSCs positive for desmin, glial fibrillary acidic protein (GFAP), and α-smooth muscle actin (α-SMA). Further, morphometric analysis showed a progressive correlation between the amount of collagen deposition and the extent of PSC activation.6 Multiple studies have established that PSCs are the principal regulator of pancreatic cancer-associated desmoplasia, promoting cancer cell growth, metastases and chemoresistance.4, 7 The terms ‘PSC’ and ‘cancer-associated fibroblast’ (CAF) have been used alternatively; however, several studies have shown that CAFs comprise a heterogeneous population of cells with potentially different origins, possibly including PSCs from the pancreas itself.8, 9, 10 It has also been shown that the majority of CAFs express fibroblast-activated protein (FAP), whereas α-SMA is co-expressed by only a subset of these cells.11, 12 Recently, Ohlund et al10 clearly demonstrated the existence of two mutually exclusive and reversible sub-types of CAFs in PDAC tissues. In addition to CAFs or myofibroblast-like cells, PDAC stroma contains large numbers of immunosuppressive immune cells such as tumor-associated macrophages, myeloid-derived suppressor cells, and regulatory T cells.13
The non-cellular component of pancreatic tumors consists largely of various extracellular matrix (ECM) proteins, proteoglycans, and glycoaminoglycans. In PDAC, the ECM is composed of the basement membrane or basal lamina and interstitial matrix. The basal lamina is prominent around duct-like structures of the cancer cells in well-differentiated PDAC. In poorly differentiated cancer or invasive forms of cancer, basal lamina structures are lost or disrupted.14 Collagen type IV and laminin are two major components of the basal lamina.15 Laminins are major ECM components required for basal lamina formation in vitro.16 PDAC is characterized by excessive deposition of ECM proteoglycans and glycoaminoglycans in the interstitial space of the tumor (interstitial matrix). The composition and organization of the tumor matrix changes dynamically during tumor progression. Collagen and fibronectin comprise the major proteins of the interstitial matrix, which also contains anti-adhesive proteins such as tenascin and vitronectin.17, 18 In addition to ECM proteins, the tumor interstitial matrix includes non-protein molecules such as hyaluronic acid (HA) or hyaluronan, a large linear glycosaminoglycan that is reportedly overexpressed in PDAC.19 Pancreatic cancer stroma also contains growth factors, enzymes, and cytokines, which directly or indirectly contribute to fibro-inflammatory reactions in PDAC.20
In pancreatic cancer research, the roles of cancer-associated fibroblasts and the extracellular matrix are complex and remain somewhat controversial. Studies have shown that deposition of excessive ECM creates a physical barrier between cancer cells and blood vessels, thus reducing effective drug delivery to the tumors. Moreover, molecular cross-talks between PSCs and pancreatic cancer cells regulate each cell type’s survival, proliferation, migration, and other protumorigenic properties.21, 22, 23 However, in recent years, experimental evidence has suggested that depletion of cancer-associated stroma aggravates PDAC.24, 25, 26 These discrepancies might be due to differences in experimental models or approaches adopted by different investigators. Importantly, growing evidence for the heterogeneity of CAFs has raised concerns about the type of stromal cells that have been targeted in different studies and their overall effect on cancer progression.9, 11 Many reviews have examined the various experimental models available to study pancreatic cancer;27 however, detailed information specifically on various models for studying pancreatic cancer-associated fibrosis is not yet available. Here, we review various in vitro and in vivo models that have been used to investigate pancreatic cancer-associated fibrosis and discuss their strengths and limitations.
In vitro models used to study desmoplasia in pancreatic cancer
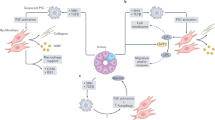
In vitro models offer the advantage of directly observing and manipulating experimental conditions. Pancreatic stellate cells or myofibroblast-like cells isolated from non-malignant or malignant pancreatic tissues have been extensively used in various in vitro studies (Figure 1; Table 1). According to the literature, PSCs isolated from multiple animal species have been used to address questions associated with pancreatic cancer desmoplasia. In certain instances, the isolated cells have been successfully immortalized to generate pancreatic stellate cell lines.

Isolation and use of PSCs/CAFs for pancreatic cancer desmoplasia studies. (a) Pancreatic stellate cells (PSCs) from wild type (wt), genetically engineered mice (GEM), and human pancreatic tissues are isolated either by density gradient centrifugation or outgrowth method. Before gradient centrifugation method, the isolated tissues are mechanically and/or enzymatically dissociated into single cell suspension. The suspension is mixed with a combination of two solutions with different viscosity (BSA and histodenz/nycodenz) and subjected to gradient centrifugation. After gradient centrifugation, PSCs appear in a fuzzy layer at the interface of these two different solutions. The cells present in the fuzzy layer (mostly quiescent PSCs/qPSCs) are further cultured and used. However, in outgrowth method, the isolated tissues (mostly pancreatic tumor tissues) are cut into small pieces and directly cultured. After few days of culture, the cancer-associated fibroblast cells (CAFs) outgrow from the tissues and spread on to the culture dish. (b) The PSCs/CAFs were then immortalized with ectopic expression of SV40 large T antigen and/or hTERT. The activated pancreatic stellate cells (aPSCs) or CAFs can further be genetically or chemically modified to identify candidates that could suppress the activation and/or growth and/or fibrogenic property of these cells. (c) To understand the interaction of PSCs/CAFs and pancreatic cancer cells (PCCs), direct and indirect co-culture methods can be performed. In the direct co-culture method both cell types are cultured in the same plate/dish. Thus both the cell types are in physical contact. In the indirect co-culture method, one cell type is cultured with the conditioned medium (CM) obtained from the other cell type or the cell types are separated by a porous physical barrier (transwell co-culture).
Isolation, Characterization and Immortalization of Primary PSCs
In most published studies, PSCs were isolated from normal pancreas, pancreatitis tissues or pancreatic cancer tissues either by density gradient centrifugation or the outgrowth method. The method for isolation of quiescent PSCs from rat pancreas using Nycodenz-based density gradient centrifugation was first developed by Apte et al.28 Vitamin A autofluorescence determination and immunocytochemical staining for desmin and GFAP were performed to assess purity of these cells. To date, density gradient centrifugation is the most commonly used method to harvest PSCs from experimental animals. Prior to centrifugation, the tissues are first treated with an enzyme cocktail for better separation of cells from the pancreatic tissues (Figure 1). PSCs isolated by this method are usually free of acinar cell, macrophage, and endothelial cell contamination. Using a similar approach, Vonlaufen et al29 isolated quiescent normal human PSCs (NhPSCs) from pancreatic tissues and verified the expression of PSC markers such as α-SMA, GFAP, desmin, TLR4, and CD14 by immunoblotting and immunocytochemistry. Isolated NhPSCs express the same markers as rat PSCs and human cancer-associated PSCs, whereas their response to PDGF and TGFβ was more similar to that of rat PSCs. In a recent study, Zhao et al reported a modified method for enzymatic digestion of pancreatic tissue prior to the density gradient centrifugation step. In this method, the investigators injected an enzyme solution into the pancreatic duct of an anesthetized rat before removing the pancreas and observed a better yield of PSCs.30 This study suggested that any method avoiding over-digestion or incomplete digestion of the pancreas before gradient centrifugation enhances PSC yield and viability.
Bachem and colleagues were the first to establish the outgrowth method of PSC isolation,31 a commonly used approach to isolate cancer-associated fibroblast and/or stellate cells from surgically removed tissues (Figure 1). In this method, fibroblast-like cells migrate/outgrow from small fragments of tissue placed in the culture medium. Using this protocol, different investigators have isolated PSCs from both cancerous and non-cancerous tissues.7, 32 The typical morphological features and expression of various PSC markers (α-SMA and/or GFAP) were assessed to check the purity of isolated cells.7, 32 Occasionally, cytokeratin expression was also checked to rule out contamination of epithelial cells.7 Compared to the density gradient centrifugation method, the outgrowth technique takes more time to obtain the desired number of cells. In the outgrowth method, firm attachment of tissue blocks to culture dishes is one of the most important steps to facilitate good outgrowth of cells. To achieve this, different investigators have adapted techniques such as initial seeding of tissue blocks in low volume culture medium to help tissues attach to the surface.33 As reported by Han et al,34 better results are obtained by seeding tissue blocks on 10 mM CaCl2/FBS pre-coated culture plates. Zhao et al30 have adopted an innovative way to accelerate the outgrowth of cancer-associated PSCs. They showed that when cancer tissue pieces were covered with plasma coagulant obtained from the same patient, the tissue blocks firmly adhered to culture plates and cells grew faster compared to controls.
Primary PSCs have a finite lifespan, and frequent isolation of primary PSCs from tissue is a time-consuming and expensive procedure. Moreover, multiple passages of cultured PSCs result in alteration of their phenotype. Therefore, efforts have been made to generate immortalized PSC cell lines (Table 1). Jesnowski et al33 immortalized human PSCs by transfection with SV40 large T antigen and human telomerase reverse transcriptase (hTERT). The immortalized PSC line RLT-PSC retains expression of α-SMA, vimentin, desmin, and GFAP. Interestingly, culture of RLT-PSC cells on matrigel followed by treatment with N-acetyl cysteine (NAC) results in deactivation of these cells.33 Similarly, Hwang et al35 have also immortalized human PSCs (hPSCs) with telomerase and SV40 large T antigen. To avoid difficulty in maintaining primary PSCs in culture, Rosendahl et al36 have generated the first conditionally immortalized human tumor-derived (TPSC) and non-tumor (NPSC) PSCs. Spontaneous immortalization of rat PSCs in vitro has also been instrumental in generating continuous PSC cell lines, which retain both morphological and molecular characteristics of primary in-culture-activated PSCs.37 An immortalized cell line has the advantage of a faster rate of division, does not undergo senescence and can be genetically manipulated for prolonged studies; however, its continuous proliferation and high growth rate do not mimic the actual phenotype of PSCs in situ. Moreover, studies carried out with a single cell line do not reproduce the variations observed in primary cells obtained from different individuals.
Utilization of PSCs in Cell Culture-Based Studies
Isolation of PSCs from rodent and human pancreas and their activation in culture have been extensively studied to understand the pathophysiological properties of these cells. Gene and protein expression analysis of activated and quiescent PSCs has led to a better understanding of the molecules involved in PSC activation and identification of molecular mediators of desmoplastic reactions in PDAC. Importantly, experimental manipulation of culture conditions and selective genetic alteration of PSCs in vitro have further improved our understanding of the mechanisms and functional involvement of these cells in PDAC. Cell culture of PSCs has also been helpful to understand their role in ECM deposition and remodeling.38 Though PSCs isolated from different sources express common molecular markers such as α-SMA, their normal culture properties and response to exogenous factors may vary. In one of our recent studies, we reported that PSCs of hamster origin express multiple proteins similar to human PSCs; however, these cells are more susceptible to disulfiram-mediated cell death than are human PSCs.39 In another study, a cross-species analysis of nicotine-induced proteomic alterations in PSCs from human, rat and mouse revealed that among all proteins identified (nicotine-induced and un-induced), 50% of proteins were common to all three species, whereas 75% of proteins were common to at least two of the three species.40 However, even within the same species, the composition and biological properties of secretory factors produced by PSCs of normal and malignant pancreas were different from each other.41 These experimental studies clearly suggest that PSCs of different origins might not always be functionally similar due to their intrinsic genetic differences and/or different culture methods.
To replicate the in vivo conditions, cancer cells and PSCs have been co-cultured in several studies. These in vitro studies have improved our understanding of the molecular crosstalk between pancreatic cancer cells and PSCs. Co-culture experiments were carried out using direct or indirect culture methods (Figure 1). In cases with direct co-culturing, cancer cells and PSCs remain in physical contact with each other, whereas for indirect co-culturing, conditioned medium obtained from one cell type was added to the other cell type or both cell types cultured using transwell inserts, where cell types remain physically separated (Figure 1).
Direct co-culture methods have been helpful in identifying mediators of molecular crosstalk between PSCs and pancreatic cancer cells and in studying their effects on activation, survival and/or proliferation of these cells.23, 42 Under a bright field microscope, the co-cultured cancer cells and PSCs can be visually distinguished based on their morphology; however, to accurately quantify the number of viable cancer cells or PSCs, cells should be labeled with a reporter system such as luciferase.39 Similarly, the use of differentially labeled pancreatic cancer cells and/or PSCs also aids in distinguishing or quantifying both cell types separately.23 To quantify the overall growth of cancer cells, investigators cultured small numbers of cancer cells (un-labeled) on a monolayer of PSCs, and the numbers of cancer cell colonies were counted.43 However, due to the presence of both cancer cells and PSCs on the same culture plate surface, direct co-culture methods are not suitable for investigating the migratory properties of individual cells. In contrast, indirect co-culturing has been frequently utilized to study the effect of secretory factors produced by both PSCs and pancreatic cancer cells on each other’s migration.44, 45
In an indirect co-culture method, cell labeling is not required to identify the individual cell types. Wu et al21 cultured AsPC-1 and BxPC-3 cells in the presence of PSC-conditioned medium and observed induction of cancer cell proliferation. Using transwell inserts, Lohr et al22 demonstrated that co-culture of fibroblasts with pancreatic cancer cells overexpressing TGF-β-induced proliferation of both cancer and fibroblast cells. As discussed above, the major drawback of the indirect co-culture method is the lack of direct physical contact between PSCs and cancer cells, which hinders multiple juxtacrine signaling that actually exists in the pancreatic tumor microenvironment. The effect of cancer cells and PSCs upon each other depends on the type of co-culture method. For example, Fujita et al23 observed increased numbers of pancreatic cancer cells with enhanced proliferation in the direct co-culture method compared to indirect co-culture. Hence, incorporation of both direct and indirect co-culture methods enables investigators to obtain a more comprehensive model of PSCs and pancreatic cancer cells effects upon each other.
Use of Non-PSCs to Delineate Crosstalk Between Stromal Cells and Cancer Cells In Vitro
In addition to PSCs, fibroblast cells of non-pancreatic origin have also been used to investigate the crosstalk between pancreatic cancer cells and fibroblasts. Using the NIH-3T3 cell line (mouse embryonic fibroblasts) stably transfected with a GLI-responsive firefly luciferase reporter (NIH-3T3/GLI-luc), Spivak-Kroizman et al46 demonstrated that activation of HIF-1α in pancreatic cancer cells strongly induces secretion of sonic hedgehog ligand (SHH) in cancer cells, which in turn stimulates stromal fibroblast cells to express more ECM proteins. Similarly, Lohr et al22 used skin fibroblast cell lines to investigate the effect of pancreatic cancer cell-derived secretory factors on fibroblast proliferation. When fibroblasts were incubated with conditioned medium derived from PANC-1/TGF-β1 cells, they showed increased expression of collagen type I protein and tyrosine phosphorylation.22 In another study, Damhofer et al47 co-cultured human pancreatic cancer cells and mouse fibroblasts (C3H10T1/2) under non-adherent low serum conditions. The tumor cells and fibroblasts (with and without HH-pathway reporter) formed mixed aggregates, mimicking in vivo findings. These aggregates were subsequently cultured for 5 days prior to further analysis. Authors selected C3H10T1/2 fibroblasts because they are highly responsive to HH ligands. Using this model, they identified multiple SHH-dependent stromal factors that contribute to pancreatic cancer pathogenesis.47 To understand the crosstalk between stromal and cancer cells, Majety et al48 co-cultured pancreatic cancer cells with MRC5 fibroblasts originating from human lung tissue. The study reported improved survival of cancer cells in the presence of these fibroblasts. Moreover, Ohuchida et al49 showed that soluble factors produced by irradiated MRC5 and pancreatic fibroblasts increased the invasiveness of pancreatic cancer cells, which was completely blocked by addition of NK4, a specific antagonist of HGF. Although fibroblasts from non-pancreatic origins have been useful for understanding the crosstalk between stromal and cancer cells, the intrinsic gene expression difference between these cells and PSCs cannot be overlooked. For instance, genome-wide assessment of gene expression analysis has shown differences between PSCs and skin fibroblasts.50 Hence, further global proteomic comparisons should be undertaken to understand the relative similarities between pancreas-associated fibroblasts or PSCs and fibroblasts from other organs.
Three-Dimensional Culture Models of Pancreatic Cancer-Associated Desmoplasia
Three-dimensional (3D) culture models are becoming increasingly popular over conventional two-dimensional (2D) cell culture due to improved tissue organization, cell–cell interactions and cell–ECM interactions that resemble in vivo architecture.51 Early 3D cultures using only pancreatic cancer cells in an ECM scaffold (cancer cell spheroids) have progressively evolved into an organotypic culture comprising multiple cell types. Three-dimensional cell culture models mimic PDAC features both at the cellular and extracellular levels. Various 3D cell culture models have been instrumental in understanding signaling between stromal and cancer cells, which can be further used to evaluate new therapeutic strategies against PDAC. Coleman et al51 have extensively discussed the evolution of 3D organotypic models of PDAC. In this review, we highlighted recent advances in the field of PDAC 3D culture models that have not been previously discussed.
Using an efficient 3D microfluidic device, Drifka et al52 developed an in vitro model of PDAC, consisting of stromal cells and cancer cells in a spatially defined 3D configuration that closely mimics the in vivo tumor microenvironment of human PDAC. The human pancreatic cancer cell line PANC-1 and PSCs obtained from pancreatic cancer patient tumor tissue were used in this study. In this model, the 3D ECM network was formulated using a hybrid of Col-I and HA.52 The authors believe that this model will be critical in elucidating the mechanisms behind cell–ECM interaction and evaluating therapeutics designed to target cellular and/or ECM components of PDAC. Recently, several investigators modified the 3D organotypic assay by culturing pancreatic cancer cells, PSCs and additional cell types together. Karnevi et al53 and Di Maggio et al54 recently cultured M2-like macrophages and HUVECs with cancer cells and PSCs in 3D organotypic culture, respectively. The presence of multicellular components in these models offers added advantages by mimicking the heterotypic composition of PDAC tissues. The ability to culture different combinations of cells (such as pancreatic cancer cells with HUVECs or PSCs with HUVECs) has enabled us to understand the contribution of individual cell types to a particular event, which would have been otherwise difficult to address in vivo. Ohlund et al studied the interaction between pancreatic cancer cells and PSCs and established a novel 3D culture model using GFP-labeled tumor-derived murine pancreatic organoids and mCherry-labeled murine PSCs. This model reproduces the desmoplastic reaction of PDAC in vitro, demonstrating the cooperative interactions between PSCs and cancer cells.10 In various 3D culture models, the use of exogenous ECM has always been a limitation in quantifying the level of ECM production by PSCs or pancreatic cancer cells. Moreover, the contribution of endogenously produced ECM by each cell type cannot be addressed in these models. To avoid the use of exogenous ECM, Ware et al55 have adopted a modified hanging drop technique to form PDAC stroma-rich spheroids (PDAC-SS) by culturing PSCs and pancreatic cancer cells in a mixture of methyl-cellulose (0.24%) and cell-specific culture medium. In another approach, Brancato et al generated PDAC microtissue by co-culturing pancreatic cancer cells (PT45) and normal fibroblast or CAFs within biodegradable microcarriers (gelatin microspheres). In this model, the gelatin scaffold degrades after a few days in culture, and all cells are surrounded by endogenous ECM produced by CAFs and/or cancer cells.56 Hence, this model appears to be a suitable tool to investigate the actual crosstalk between cancer cells and stroma.
To investigate PDAC pathogenesis, Boj et al57 recently characterized a neoplastic pancreatic 3D organoid model. The authors initially generated murine primary tumors (mT) and metastatic (mM) organoids by harvesting tissues from genetically engineered mouse models (GEMMs) of pancreatic cancer, followed by enzymatic digestion and seeding in growth factor-reduced matrigel. Adopting similar methods, tumor organoids were also established from freshly resected human PDAC tissues (hT organoids).57 The authors reported that hT organoids can be cryopreserved and passaged indefinitely. These organoids (both mouse and human origin) were further orthotopically implanted in Nu/Nu mice, and organoid-derived tumors showed deposition of collagen-rich stroma and recruitment of αSMA-positive cells.
In vivo models
Co-implantation of pancreatic cancer cells and PSCs or implantation of only cancer cells in heterologous or homologous hosts has been instrumental in understanding pancreatic cancer desmoplasia and evaluating novel therapeutics against it. Nevertheless, pancreas-specific genetic modifications in mice were critical experimental tools for understanding molecular mechanisms associated with desmoplastic reactions in PDAC. These animal models have also been useful in evaluating target-specific therapeutic interventions against PDAC.
Implantation of Pancreatic Cancer Cells and/or PSCs
Experimental pancreatic tumors are established by implanting human or murine cancer cells in immunodeficient (xenograft models) or immunocompetent (syngeneic models) animals. The cells can be injected at ectopic or orthotopic sites, and injection may consist of only cancer cells or a combination of cancer cells and PSCs. To generate desmoplastic tumors, cancer cells that are capable of attracting host fibrogenic cells are injected alone, and cancer cells that are either not capable or less capable of attracting host fibrogenic cells are co-injected with isolated PSCs. For efficient tumor uptake, in certain cases, the cells are mixed with extracellular matrix proteins (matrigel) and injected into animals. To reproduce the actual tumor microenvironment, patient-derived cancer tissue can be directly used in xenograft models (patient-derived xenograft or PDX models). All these models have inherent strengths and limitations (Table 2), and the selection of a model depends primarily on the question being addressed and the nature of the cancer cells being injected.
Subcutaneous Implantation
To generate desmoplastic pancreatic tumors, Kozono et al subcutaneously co-injected pancreatic cancer cells (SUIT-2) and human PSCs. Resulting tumors showed marked desmoplasia with larger Sirius red-positive areas and more α-SMA-positive cells than tumors consisting of SUIT-2 cells alone.7 Using this model, the authors demonstrated that pirfenidone suppresses growth of PSCs, inhibiting tumor formation by SUIT-2 cancer cells.7 In another study, co-injection of serine protease inhibitor Nexin 2 (SERPINE2)-expressing SUIT-2 cells with PSCs in a xenograft model established the role of PSCs in potentiating the pro-invasive effects of SERPINE2 expression in pancreatic cancer cells.58 Similarly, co-injection of AsPC-1 cells with PSCs in athymic nude mice resulted in the formation of larger subcutaneous tumors than those obtained from injection of AsPC-1 cells alone, as reported with other pancreatic cancer cell lines.59 Further, the AsPC-1 and PSC co-injection model has been used to demonstrate the effect of olmesartan, an angiotensin II type I receptor blocker, in inhibiting tumor growth by targeting stellate cell activities.59
Orthotopic Implantation
Crosstalk between pancreatic cancer cells and PSCs has been further characterized in vivo by injecting cancer cells, with or without PSCs, directly into the pancreas of animals. Co-injection of hPSCs and human pancreatic cancer cells (MiaPaCa-2 and PANC-1) in orthotopic nude mice resulted in increased tumor size with regional and distant metastasis compared to cancer cells alone, while no tumors were observed in the mice injected with hPSCs alone.45 Tumors generated through hPSC and cancer cell co-injection exhibited increased desmoplastic lesions.45 Likewise, Hwang et al35 studied the role of cancer-associated stromal fibroblasts in pancreatic cancer progression by orthotopic co-injection of immortalized hPSCs and BxPC-3 cancer cells, resulting in increased primary tumor incidence, size and metastasis. Notably, the ratio of PSCs to cancer cells injected into the pancreas influenced the level of desmoplasia, tumor growth and incidence of metastasis.35 To understand the role of PSCs in pancreatic cancer cell metastasis, Xu et al60 co-injected cancer cells (AsPC-1) with PSCs isolated from two different sources: cancer-associated PSCs (CAhPSCs) and normal PSCs (NhPSCs). They observed that mice injected with AsPC-1 and CAhPSCs/NhPSCs showed early tumor incidence and increased tumor size. Notably, mice injected with CAhPSCs/NhPSCs alone did not develop tumors, suggesting that PSCs themselves are not tumorigenic. H&E stained tumor sections showed visible bands of fibrosis in primary tumors of mice injected with AsPC-1 plus CAhPSCs/NhPSCs, which resembled the desmoplasia observed in human pancreatic cancer. To detect the presence of migratory hPSCs at sites of metastasis, a sex mismatch study was carried out using female mice implanted with female cancer cells with/without male hPSCs in their pancreas. The presence of the Y chromosome in the metastatic nodules of mice injected with AsPC-1 and CAhPSCs/NhPSCs cells confirmed the presence of exogenously introduced male hPSCs.60
Certain human pancreatic cancer cell lines are known to generate fibrotic pancreatic tumors by themselves (without co-injection of PSCs). These cancer cells secrete factors that induce recruitment of host fibrogenic cells to the tumor site. Lohr et al22 reported induction of stroma in nude mice following orthotopic injection of PANC-1 cells overexpressing TGF-β1. Similarly, injection of Capan-2 cancer cell lines by themselves in athymic mice generated desmoplastic tumors.61 When these mice were treated with 5E1, a blocking antibody for SHH, there was a significant decrease in the degree of desmoplasia.61 Primary xenografts established through orthotopic implantation of tumor tissue also generated fibrotic tumors by recruiting host stromal cells. Chang et al62 investigated the effect of hedgehog pathway inhibition on tumor-stroma crosstalk using two mouse models designated Ontario Cancer Institute Pancreas 19 (OCIP19), constituting dense fibrotic stroma with infiltration of α-SMA-positive cells, and OCIP23, containing less stroma and fewer α-SMA-positive cells. Analysis of 5E1 treatment showed no significant decrease in collagen or α-SMA expression in both the models, whereas a reduction in tumor growth was evident and was even more pronounced in OCIP19 mice.62
Non-human pancreatic cancer cell lines have also been very useful in generating pancreatic orthotopic tumors with desmoplasia. To understand the effect of metformin on the desmoplastic reactions observed in obese/diabetic individuals with pancreatic cancer, Incio et al63 used two immunocompetent syngeneic mouse models. To induce pancreatic tumors in diabetic mice, wild type C57BL/6 and FVB male mice were fed with a high fat diet and injected with PAN02 and AK4.4 cells, respectively. Orthotopic pancreatic tumors were generated by implanting a small piece of viable tumor tissue obtained from a separate donor into FVB (AK4.4 model) and C57BL/6 (PAN02 model) recipient mice. Reduced M2 polarization and tumor-associated macrophage infiltration were evident in both models. In the AK4.4 model, metformin reduced hyaluronan and collagen-I expression by directly suppressing TGF-β signaling in PSCs. In the PAN02 model (less desmoplastic), though the reduction in hyaluronan and collagen level was not very significant, metformin treatment was still able to decrease the density of activated PSCs.63 Sherman et al used p53 2.1.1 mouse pancreatic cancer cell lines derived from P48-Cre;LSL-KrasG12D/+;p53lox/+ mice to generate an orthotopic allograft model. The authors observed measurable PSC activation accompanied by fibrosis in allograft recipients. Further, calcipotriol treatment decreased stromal activation and enhanced gemcitabine efficacy in vivo.64 In another study, Sousa et al established a syngeneic orthotopic tumor by co-injecting mouse PSCs and cancer cells obtained from KrasG12D, p53 L/+ mice. In this model, murine PSCs enhanced tumor engraftment in an autophagy-dependent manner.65 Recently, we characterized a hamster pancreatic cancer cell line (HapT1)-based syngeneic orthotopic tumor model (Figure 2). Our detailed analysis indicated that HapT1 orthotopic tumors harbor myofibroblast-like cells and show extensive deposition of ECM proteins such as collagen and fibronectin.39

HapT1-derived homologous tumors as a model of pancreatic cancer desmoplasia. Injection of hamster pancreatic cancer cells, HapT1 into the pancreas of Syrian golden hamster (Mesocricetus auratus; homologous host) generates predictable tumors in a short period of time (a and b). Further, trichome staining and immunohistochemistry for α-SMA in HapT1 tumor sections shows deposition of collagen (c; blue) and presence of myofibroblast-like cells in the tumor matrix (d; brown), respectively. A full color version of this figure is available at the Laboratory Investigation journal online.
Genetically Engineered Mouse Models
GEMMs have been very useful in understanding the pathophysiology of PDAC. With different combinations of genetic manipulations, investigators have studied the role of genetic factors in the manifestation of particular PDAC phenotypes. Different GEMMs of pancreatic cancer have been used to examine the crosstalk between cancer cells and its stroma and evaluate therapeutic strategies against cancer-associated stroma. To assess the role of mutated Kras in pancreatic tumorigenesis, Hingorani et al directed endogenous KrasG12D expression to the progenitor cells of mouse pancreas by interbreeding LSL-KrasG12D mice with mice expressing Cre recombinase specific to the pancreatic promoter (PDX-1 or P48). The compound strains carrying either LSL-KrasG12D;PDX-1-Cre; or LSL-KrasG12D;P48-Cre develop a full spectrum of PanIN lesions that were histologically similar to those seen in human patients with highly invasive and metastatic phenotypes.66 In the presence of various exogenous factors, these compound strains showed more robust fibro-inflammatory reactions. When 10-week-old LSL-KrasG12D;PDX-1-Cre offspring were exposed to cigarette smoke for 20 weeks, formation of PanIN lesions accelerated and PSC activation was stimulated, correlating with pancreatic fibrosis.67 In another study, when 6-week-old LSL-KrasG12D; P48-Cre mice were treated with cerulein or the TLR7 ligand ssRNA40, animals showed more extensive fibro-inflammatory stromal growth.68 Addition of further genetic modifications along with mutated Kras (LSL-KrasG12D/+;LSL-Trp53R172H/+;Pdx-1-Cre) has also resulted in the formation of fibrotic pancreatic tumors in mice.69 Likewise, in the KPGC model (KrasLSL-G12D/+;Trp53LSL-R172H/+;R26LSL-GFP/+;Cre), the level of collagen and glycosaminoglycan (GAG) deposition increased during progression of pancreatic cancer.70 Particularly, HA was excessively deposited from early precursor lesions.
TGF-α overexpression in mouse pancreas (TGF-α transgenic mice) is known to induce extensive intra-pancreatic fibrosis and matrix accumulation.71 Moreover, various pancreas-specific genetic alterations that activate TGF-β signaling also produce pancreatic tumors with desmoplastic lesion. Expression of Snail in EL-KrasG12D/El-tTA+/TRE-Snail+ transgenic mice increased collagen production in PSCs via increased TGF-β signaling.72 Similarly, Krantz et al73 established another model, EL-KrasGD12+/El-tTA+/TRE-MT1-MMP+ mice, in which both activated KrasG12D and MT1-MMP (membrane type-1 matrix metalloproteinase) were expressed in the pancreas. These mice developed dysplastic, intra-ductal papillary mucinous neoplasms (IPMNs) with increased fibrosis (increased collagen production by PSCs) associated with increased Smad2 phosphorylation, as evidenced by increased TGF-β signaling.73 Ijichi et al74 used pancreas epithelium-specific TGF-β receptor type II (Tgfbr2) knockout mice in the context of Kras activation (Kras+Tgfbr2KO) as a model of PDAC to study tumor-stromal interaction. PDAC tissues in these mice revealed abundant stromal components and strong expression of connective tissue growth factor (Ctgf) in the tumor-stromal border, suggesting an active tumor-stromal interaction.74 In another study, Ozdemir et al24 crossed Ptf1acre/+;LSL-KrasG12D/+;Tgfbr2flox/flox (PKT) mice with α-SMA-tk transgenic mice (α-SMA-tk) to selectively target proliferating α-SMA+ myofibroblasts following treatment with systemic ganciclovir. This study demonstrated that depletion of α-SMA+ myofibroblasts results in multiple adverse events, ultimately leading to accelerated disease progression and decreased survival.
Some GEMMs have been used to evaluate the therapeutic potential of drugs targeting tumor stroma in combination with chemotherapeutics. In the KPC model, IPI-926, a drug that depletes tumor stroma by inhibiting the Hh signaling pathway, also enhances intra-tumor delivery of gemcitabine.75 These experimental data suggest that depletion of pancreatic cancer stroma augments tumor angiogenesis, enhancing drug delivery. The KPC model has also been instrumental in evaluating the efficacy of calcipotriol and gemcitabine combination therapy, which significantly reduced tumor volume and the number of tumor-associated fibroblasts.64 Using KPC mice, a recent study has shown the role of CAFs in scavenging gemcitabine and their contribution to chemoresistance in PDAC.76 Provenzano et al70 investigated the therapeutic efficacy of PEGPH20, which ablates HA in PDA tissue and normalizes IFP (interstitial fluid pressure) in the KPGC model. In this study, PEGPH20 and gemcitabine co-administration permanently altered the stroma and increased the overall survival of mice.70 As fibrosis contributes to chemoresistance in PDAC, Masso-Valles et al77 conducted a study to investigate the anti-fibrotic effect of ibrutinib in PDAC treatment. Experiments carried out in p53ER/ER;LSL-KRasG12D;Pdx-1-Cre mice demonstrated that ibrutinib prevents mast cell degranulation and collagen deposition. Notably, the combined therapy with ibrutinib and gemcitabine conferred a better survival advantage to the treated mice compared to gemcitabine alone.77 Ijichi et al74 showed that treatment of Kras+Tgfbr2KO mice with a CXCR2 inhibitor suppressed tumor progression. Thus, targeting the tumor-stroma interaction is a promising therapeutic strategy against PDAC.
Conclusions
The use of various in vitro and in vivo experimental models for pancreatic cancer-associated fibrosis studies are rapidly evolving. The development and refinement of PSC isolation methods and culture techniques are considered major technical achievements in this field. Recent advancements in 3D culture models have added a new dimension and significance to the field. GEMMs have provided the most clinically relevant tool to understand pancreatic cancer-associated desmoplasia. In the aforementioned experimental models, most of the in vitro and in vivo models used PSCs/CAFs isolated from rodent or human patients. Despite their utility, certain important factors should be considered when using these models: (1) In-culture activation of PSCs is believed to be a consequence of culture stress; hence, the molecular and/or cellular properties of these cells may not exactly mimic activated PSCs/CAFs present in PDAC tissues. (2) In-culture-activated PSCs of different passage numbers have different growth rates and express variable levels of ECM proteins; therefore, in pancreatic cancer cell and PSC co-injection experiments, PSCs of the same passage number should be used in all groups of animals (better if isolated from the same animal or human patient). (3) Some cancer cell lines need to be injected with ECM proteins/matrigel to generate experimental tumors; as a result, quantification of ECM proteins in these tumors may not reflect ECM produced by stromal cells and/or cancer cells. (4) Many orthotopically grown pancreatic tumors exhibit a dense fibrotic reaction at the periphery of the tumor mass or at the border region of normal and cancer tissues (capsule-like structure); however, there might be little or no desmoplastic reaction in the actual tumor parenchyma. A few other limitations associated with different types of animals models used in pancreatic cancer desmoplasia studies are mentioned in Table 2.
Myofibroblast-like cells present in pancreatic cancer tissue might originate from various other sources, including (i) recruitment of pre-existing stromal/fibroblast cells, (ii) transdifferentiation of mesenchymal stem cells, and (iii) epithelial-to-mesenchymal transition of cancer cells.78, 79 Unfortunately, over the last few years, emphasis on PSCs has overshadowed the significance of myofibroblast-like cells having potentially different origins. Recent experimental evidence from animal models has suggested that tumor-associated fibroblasts might suppress rather than promote tumor growth.24 Therefore, it is critical to investigate the complexity of the tumor-stroma relationship and to understand the role of myofibroblast-like cells that arise from divergent sources. Efforts should be made to develop specific markers to identify different sub-types of myofibroblast-like cells to aid in quantifying the proportion of different subsets of these cells in different animal models of pancreatic cancer. Essentially, this knowledge will also help in correlating the outcome of therapeutic agents with the type of myofibroblast-like cells present in a tumor.
References
Siegel RL, Miller KD, Jemal A . Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30.
Ferlay J, Soerjomataram I, Dikshit R et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 2015;136:E359–E386.
Rasheed ZA, Matsui W, Maitra A. Pathology of pancreatic stroma in PDAC. In: Grippo PJ, Munshi HG (eds). Pancreatic Cancer and Tumor Microenvironment. Transworld Research Network: Trivandrum, India, 2012.
Schober M, Jesenofsky R, Faissner R et al. Desmoplasia and chemoresistance in pancreatic cancer. Cancers 2014;6:2137–2154.
Apte MV, Pirola RC, Wilson JS . Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol 2012;3:344.
Apte MV, Park S, Phillips PA et al. Desmoplastic reaction in pancreatic cancer: role of pancreatic stellate cells. Pancreas 2004;29:179–187.
Kozono S, Ohuchida K, Eguchi D et al. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res 2013;73:2345–2356.
Augsten M . Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol 2014;4:62.
Sugimoto H, Mundel TM, Kieran MW et al. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther 2006;5:1640–1646.
Ohlund D, Handly-Santana A, Biffi G et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med 2017;214:579–596.
Jacob M, Chang L, Pure E . Fibroblast activation protein in remodeling tissues. Curr Mol Med 2012;12:1220–1243.
Kalluri R, Zeisberg M . Fibroblasts in cancer. Nat Rev Cancer 2006;6:392–401.
Clark CE, Hingorani SR, Mick R et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res 2007;67:9518–9527.
Ingber DE, Madri JA, Jamieson JD . Role of basal lamina in neoplastic disorganization of tissue architecture. Proc Natl Acad Sci USA 1981;78:3901–3905.
van der Zee JA, van Eijck CH, Hop WC et al. Tumour basement membrane laminin expression predicts outcome following curative resection of pancreatic head cancer. Br J Cancer 2012;107:1153–1158.
Yamanari H, Suganuma T, Iwamura T et al. Extracellular matrix components regulating glandular differentiation and the formation of basal lamina of a human pancreatic cancer cell line in vitro. Exp Cell Res 1994;211:175–182.
Linder S, Castanos-Velez E, von Rosen A et al. Immunohistochemical expression of extracellular matrix proteins and adhesion molecules in pancreatic carcinoma. Hepatogastroenterology 2001;48:1321–1327.
Esposito I, Penzel R, Chaib-Harrireche M et al. Tenascin C and annexin II expression in the process of pancreatic carcinogenesis. J Pathol 2006;208:673–685.
Provenzano PP, Hingorani SR . Hyaluronan, fluid pressure, and stromal resistance in pancreas cancer. Br J Cancer 2013;108:1–8.
Mahadevan D, Von Hoff DD . Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther 2007;6:1186–1197.
Wu YS, Looi CY, Subramaniam KS et al. Soluble factors from stellate cells induce pancreatic cancer cell proliferation via Nrf2-activated metabolic reprogramming and ROS detoxification. Oncotarget 2016;7:36719–36732.
Lohr M, Schmidt C, Ringel J et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res 2001;61:550–555.
Fujita H, Ohuchida K, Mizumoto K et al. Tumor-stromal interactions with direct cell contacts enhance proliferation of human pancreatic carcinoma cells. Cancer Sci 2009;100:2309–2317.
Ozdemir BC, Pentcheva-Hoang T, Carstens JL et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014;25:719–734.
Rhim AD, Oberstein PE, Thomas DH et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014;25:735–747.
Lee JJ, Perera RM, Wang H et al. Stromal response to Hedgehog signaling restrains pancreatic cancer progression. Proc Natl Acad Sci USA 2014;111:E3091–E3100.
Herreros-Villanueva M, Hijona E, Cosme A et al. Mouse models of pancreatic cancer. World J Gastroenterol 2012;18:1286–1294.
Apte MV, Haber PS, Applegate TL et al. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut 1998;43:128–133.
Vonlaufen A, Phillips PA, Yang L et al. Isolation of quiescent human pancreatic stellate cells: a promising in vitro tool for studies of human pancreatic stellate cell biology. Pancreatology 2010;10:434–443.
Zhao L, Cai B, Lu Z et al. Modified methods for isolation of pancreatic stellate cells from human and rodent pancreas. J Biomed Res 2016;30:510–516.
Bachem MG, Schneider E, Gross H et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology 1998;115:421–432.
Bachem MG, Schunemann M, Ramadani M et al. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005;128:907–921.
Jesnowski R, Furst D, Ringel J et al. Immortalization of pancreatic stellate cells as an in vitro model of pancreatic fibrosis: deactivation is induced by matrigel and N-acetylcysteine. Lab Invest 2005;85:1276–1291.
Han S, Delitto D, Zhang D et al. Primary outgrowth cultures are a reliable source of human pancreatic stellate cells. Lab Invest 2015;95:1331–1340.
Hwang RF, Moore T, Arumugam T et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res 2008;68:918–926.
Rosendahl AH, Gundewar C, Said Hilmersson K et al. Conditionally immortalized human pancreatic stellate cell lines demonstrate enhanced proliferation and migration in response to IGF-I. Exp Cell Res 2015;330:300–310.
Masamune A, Satoh M, Kikuta K et al. Establishment and characterization of a rat pancreatic stellate cell line by spontaneous immortalization. World J Gastroenterol 2003;9:2751–2758.
Phillips PA, McCarroll JA, Park S et al. Rat pancreatic stellate cells secrete matrix metalloproteinases: implications for extracellular matrix turnover. Gut 2003;52:275–282.
Suklabaidya S, Das B, Ali SA et al. Characterization and use of HapT1-derived homologous tumors as a preclinical model to evaluate therapeutic efficacy of drugs against pancreatic tumor desmoplasia. Oncotarget 2016;7:41825–41842.
Paulo JA, Urrutia R, Kadiyala V et al. Cross-species analysis of nicotine-induced proteomic alterations in pancreatic cells. Proteomics 2013;13:1499–1512.
Duluc C, Moatassim-Billah S, Chalabi-Dchar M et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol Med 2015;7:735–753.
Froeling FE, Feig C, Chelala C et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology 2011;141:1486–1497.
Mantoni TS, Lunardi S, Al-Assar O et al. Pancreatic stellate cells radioprotect pancreatic cancer cells through beta1-integrin signaling. Cancer Res 2011;71:3453–3458.
Kikuta K, Masamune A, Watanabe T et al. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem Biophys Res Commun 2010;403:380–384.
Vonlaufen A, Joshi S, Qu C et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res 2008;68:2085–2093.
Spivak-Kroizman TR, Hostetter G, Posner R et al. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res 2013;73:3235–3247.
Damhofer H, Medema JP, Veenstra VL et al. Assessment of the stromal contribution to Sonic Hedgehog-dependent pancreatic adenocarcinoma. Mol Oncol 2013;7:1031–1042.
Majety M, Pradel LP, Gies M et al. Fibroblasts influence survival and therapeutic response in a 3D co-culture model. PLoS ONE 2015;10:e0127948.
Ohuchida K, Mizumoto K, Murakami M et al. Radiation to stromal fibroblasts increases invasiveness of pancreatic cancer cells through tumor-stromal interactions. Cancer Res 2004;64:3215–3222.
Buchholz M, Kestler HA, Holzmann K et al. Transcriptome analysis of human hepatic and pancreatic stellate cells: organ-specific variations of a common transcriptional phenotype. J Mol Med (Berl) 2005;83:795–805.
Coleman SJ, Watt J, Arumugam P et al. Pancreatic cancer organotypics: High throughput, preclinical models for pharmacological agent evaluation. World J Gastroenterol 2014;20:8471–8481.
Drifka CR, Eliceiri KW, Weber SM et al. A bioengineered heterotypic stroma-cancer microenvironment model to study pancreatic ductal adenocarcinoma. Lab Chip 2013;13:3965–3975.
Karnevi E, Rosendahl AH, Hilmersson KS et al. Impact by pancreatic stellate cells on epithelial-mesenchymal transition and pancreatic cancer cell invasion: adding a third dimension in vitro. Exp Cell Res 2016;346:206–215.
Di Maggio F, Arumugam P, Delvecchio FR et al. Pancreatic stellate cells regulate blood vessel density in the stroma of pancreatic ductal adenocarcinoma. Pancreatology 2016;16:995–1004.
Ware MJ, Keshishian V, Law JJ et al. Generation of an in vitro 3D PDAC stroma rich spheroid model. Biomaterials 2016;108:129–142.
Brancato V, Comunanza V, Imparato G et al. Bioengineered tumoral microtissues recapitulate desmoplastic reaction of pancreatic cancer. Acta Biomater 2017;49:152–166.
Boj SF, Hwang CI, Baker LA et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160:324–338.
Buchholz M, Biebl A, Neesse A et al. SERPINE2 (protease nexin I) promotes extracellular matrix production and local invasion of pancreatic tumors in vivo. Cancer Res 2003;63:4945–4951.
Masamune A, Hamada S, Kikuta K et al. The angiotensin II type I receptor blocker olmesartan inhibits the growth of pancreatic cancer by targeting stellate cell activities in mice. Scand J Gastroenterol 2013;48:602–609.
Xu Z, Vonlaufen A, Phillips PA et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol 2010;177:2585–2596.
Bailey JM, Swanson BJ, Hamada T et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res 2008;14:5995–6004.
Chang Q, Foltz WD, Chaudary N et al. Tumor-stroma interaction in orthotopic primary pancreatic cancer xenografts during hedgehog pathway inhibition. Int J Cancer 2013;133:225–234.
Incio J, Suboj P, Chin SM et al. Metformin reduces desmoplasia in pancreatic cancer by reprogramming stellate cells and tumor-associated macrophages. PLoS ONE 2015;10:e0141392.
Sherman MH, Yu RT, Engle DD et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014;159:80–93.
Sousa CM, Biancur DE, Wang X et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016;536:479–483.
Hingorani SR, Petricoin EF, Maitra A et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003;4:437–450.
Kumar S, Torres MP, Kaur S et al. Smoking accelerates pancreatic cancer progression by promoting differentiation of MDSCs and inducing HB-EGF expression in macrophages. Oncogene 2015;34:2052–2060.
Ochi A, Graffeo CS, Zambirinis CP et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J Clin Invest 2012;122:4118–4129.
Hingorani SR, Wang L, Multani AS et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005;7:469–483.
Provenzano PP, Cuevas C, Chang AE et al. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012;21:418–429.
Schmid RM, Kloppel G, Adler G et al. Acinar-ductal-carcinoma sequence in transforming growth factor-alpha transgenic mice. Ann N Y Acad Sci 1999;880:219–230.
Shields MA, Ebine K, Sahai V et al. Snail cooperates with KrasG12D to promote pancreatic fibrosis. Mol Cancer Res 2013;11:1078–1087.
Krantz SB, Shields MA, Dangi-Garimella S et al. MT1-MMP cooperates with Kras(G12D) to promote pancreatic fibrosis through increased TGF-beta signaling. Mol Cancer Res 2011;9:1294–1304.
Ijichi H, Chytil A, Gorska AE et al. Inhibiting Cxcr2 disrupts tumor-stromal interactions and improves survival in a mouse model of pancreatic ductal adenocarcinoma. J Clin Invest 2011;121:4106–4117.
Olive KP, Jacobetz MA, Davidson CJ et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009;324:1457–1461.
Hessmann E, Patzak MS, Klein L et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2017.
Masso-Valles D, Jauset T, Serrano E et al. Ibrutinib exerts potent antifibrotic and antitumor activities in mouse models of pancreatic adenocarcinoma. Cancer Res 2015;75:1675–1681.
Scarlett CJ, Colvin EK, Pinese M et al. Recruitment and activation of pancreatic stellate cells from the bone marrow in pancreatic cancer: a model of tumor-host interaction. PLoS ONE 2011;6:e26088.
Xie D, Xie K . Pancreatic cancer stromal biology and therapy. Genes Dis 2015;2:133–143.
Satoh M, Masamune A, Sakai Y et al. Establishment and characterization of a simian virus 40-immortalized rat pancreatic stellate cell line. Tohoku J Exp Med 2002;198:55–69.
Piao RL, Xiu M, Brigstock DR et al. An immortalized rat pancreatic stellate cell line RP-2 as a new cell model for evaluating pancreatic fibrosis, inflammation and immunity. Hepatobiliary Pancreat Dis Int 2015;14:651–659.
Haas SL, Fitzner B, Jaster R et al. Transforming growth factor-beta induces nerve growth factor expression in pancreatic stellate cells by activation of the ALK-5 pathway. Growth Factors 2009;27:289–299.
Froeling FE, Mirza TA, Feakins RM et al. Organotypic culture model of pancreatic cancer demonstrates that stromal cells modulate E-cadherin, beta-catenin, and Ezrin expression in tumor cells. Am J Pathol 2009;175:636–648.
Mathison A, Liebl A, Bharucha J et al. Pancreatic stellate cell models for transcriptional studies of desmoplasia-associated genes. Pancreatology 2010;10:505–516.
Hamada S, Masamune A, Yoshida N et al. IL-6/STAT3 plays a regulatory role in the interaction between pancreatic stellate cells and cancer cells. Dig Dis Sci 2016;61:1561–1571.
Acknowledgements
We sincerely acknowledge the support provided by the Institute of Life Sciences, Bhubaneswar to SBS; extramural grant by the Department of Biotechnology (DBT), Government of India to SBS (BT/PR13502/MED/30/1555/2015); Senior Research Fellowship by DBT, Government of India to SS and SERB National Post-Doctoral fellowship by the Department of Science and Technology, Government of India to PD. We thank Dr Neera Singh for editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
The role of stroma in pancreatic cancer is controversial, potentially due to the variability observed among different model systems. In this review, the authors extensively examine several original research studies related to various models currently available for pancreatic cancer desmoplasia, highlighting their strengths and weaknesses.
Rights and permissions
About this article

Cite this article
Suklabaidya, S., Dash, P., Das, B. et al. Experimental models of pancreatic cancer desmoplasia. Lab Invest 98, 27–40 (2018). https://doi.org/10.1038/labinvest.2017.127
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2017.127
This article is cited by
-
Evaluation of growth-induced, mechanical stress in solid tumors and spatial association with extracellular matrix content
Biomechanics and Modeling in Mechanobiology (2023)
-
Fluvastatin sensitizes pancreatic cancer cells toward radiation therapy and suppresses radiation- and/or TGF-β-induced tumor-associated fibrosis
Laboratory Investigation (2022)
-
Preclinical models of pancreatic ductal adenocarcinoma: challenges and opportunities in the era of precision medicine
Journal of Experimental & Clinical Cancer Research (2021)
