
Abstract
Lung epithelial apoptosis and inflammatory responses are important pathological processes in many pulmonary disorders. ResolvinD1 (RvD1), generated in inflammatory resolution processes, reduces inflammatory responses in animal models of lung diseases. The aim of this study was to investigate whether RvD1 attenuates apoptosis and proinflammatory responses in primary human alveolar epithelial type 2 cells (AEC2 cells) that are exposed to lipopolysaccharide (LPS) in vitro. We examined the percentage of apoptotic AEC2 cells by flow cytometry. The expression levels of cytokines and chemokines were determined by ELISA and microarray. The expression levels of molecular signaling modulators were evaluated by western blot. LPS-stimulated AEC2 cells pretreated with RvD1 exhibited a statistically significant reduction in apoptosis. The pretreatment of LPS-stimulated cells with RvD1 stimulated the phosphorylation of AKT and prevented the cleavage of caspase-3, the upregulation of Bax, and the downregulation of Bcl-2. The antiapoptotic effects of RvD1 were abrogated upon pretreatment with a PI3K inhibitor. In addition, RvD1 reduced the release of cytokines and chemokines, and inhibited the degradation and phosphorylation of IκB-α in LPS-stimulated AEC2 cells. RvD1 reduces apoptosis of LPS-exposed AEC2 cells by inducing the phosphorylation of AKT and attenuates the inflammatory response by suppressing the degradation and phosphorylation of IκB-α.
Similar content being viewed by others

Main
The lung epithelium has a pivotal role in the regulation of the inflammatory response and lung repair in many lung diseases. In particular, alveolar epithelial type 2 cells (AEC2 cells), which constitute the first line of immune defense by releasing a variety of inflammatory mediators, can induce the proliferation and differentiation of epithelial cells during epithelial barrier injury.1, 2 Extensive apoptosis of alveolar epithelial cells is a major outcome of the destruction of the alveolar epithelium, which results in increases in alveolar capillary permeability.3
Bacterial cell wall products (i.e., lipopolysaccharide (LPS)), which are associated with worsening symptoms in pulmonary disorders, can destroy alveolar epithelial cells and induce apoptosis in an animal model of ALI.4 In addition, the stimulation of AEC2 cells with LPS induces the production of chemokines and cytokines and the migration of leukocytes.1 Thus, a strategy that attenuates extensive apoptosis of epithelial cells and the inflammatory response induced by LPS would be beneficial for the treatment of pulmonary disorders.5
Specialized proresolving lipid mediators (SPMs), such as lipoxins, resolvins (Rvs), and protectins, are endogenously biosynthesized during the resolution phase of inflammation and exhibit potent anti-inflammatory and proresolving effects.6 ResolvinE1 and resolvinD1 (RvD1) have been shown to exhibit protective effects in an animal model of acute lung injury.7, 8 ResolvinE1 has a direct protective effect against ischemia–reperfusion injury in cardiomyocytes.9 RvD1, a D-series resolvin, is biosynthesized from DHA and exhibits potent anti-inflammatory effects in vivo.10 Some G-protein-coupled receptors for RvD1, that is, ALX/FPR2 and GPR32, have been identified. Signaling through these receptors mediates the effects of RvD1 and enhances the macrophage phagocytosis and apoptotic PMNs.11
Based on these previous studies, we determined whether RvD1 could reduce both the release of inflammatory mediators and apoptosis in LPS-stimulated AEC2 cells in vitro. Our results indicate that RvD1 promotes the survival of AEC2 cells via a mechanism that involves the phosphorylation of AKT and reduces the release of cytokines and chemokines through the inhibition of IκB-α phosphorylation and degradation.
MATERIALS AND METHODS
The following reagents were obtained from Cayman Chemical (Ann Arbor, Michigan, USA): RvD1, wortmannin, LY294002, and PD98059. BAY-11-7082 and Z-VAD were purchased from Calbiochem (San Diego, CA, USA). LPS and the ECM gel from Engelbreth–Holm–Swarm mouse sarcoma were obtained from Sigma-Aldrich (St Louis, MO, USA). Recombinant human keratinocyte growth factor was obtained from R&D Systems (Minneapolis, MN, USA). The following antibodies were obtained from Cell Signaling Technology (Danvers, MA, USA): caspase-3, Bcl-2 rabbit mAb (50E3), AKT, phospho-AKT (Ser473), IκB-α mAb (44D4) rabbit mAb, and phospho-IκB-α (Ser32) (14D4) rabbit mAb (14D4). The BAX rabbit monoclonal antibody was obtained from Epitomics (Burlingame, CA, USA). β-Actin was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Isolation and Culture of AEC2 Cells
AEC2 cells were isolated from lungs of grossly normal appearance after resection for lung carcinoma. The cells were isolated with the approval of the Ethical Committee of Tongji Medical College as described previously.1, 12, 13, 14 Briefly, the tissue was perfused and inflated with collagenase I (1% in HBSS; Sigma-Aldrich) and incubated at 37 °C for 1 h. The collagenase I was replaced two times during this incubation period. The tissue was finely chopped in the presence of newborn calf serum (Invitrogen Life Technologies, Carlsbad, CA, USA). The chopped tissue was then incubated with DNase (250 μg/ml; Invitrogen Life Technologies), and the mixture was successively passed through a 300- and 40-μm filter (BD Bioscience, San Diego, CA, USA) to remove all of the large tissue debris. The cell suspension was then centrifuged (290 g for 10 min at 20 °C), and the resulting pellet was resuspended in DMEM/F12 medium containing 50 μg/ml of DNase. The cells were isolated by filtration and partially purified by centrifugation on a discontinuous density gradient composed of OptiPrep (Axis-Shield, Dundee, Scotland, UK) with densities of 1.080 and 1.040. The cells then underwent negative selection with CD14-coated magnetic beads (Dynal Biotech, ASA, Oslo, Norway) and were bound to IgG-coated Petri dishes (Sigma-Aldrich). These cells were confluent by 48 h and thoroughly characterized using electron microscopy and the expression of surfactant protein A by immunohistochemical staining. Electron microscopy revealed that the cells had a cuboidal morphology, surfactant-containing lamellar bodies, tight junctions, and microvilli.
The isolated cells were plated on Petri dishes coated with a mixture of rat tail collagen and Matrigel (Sigma-Aldrich) in 10% FBS in DMEM and then cultured with 1% charcoal-stripped FBS with keratinocyte growth factor, isobutylmethylxanthine, 8-bromo-cAMP, dexamethasone, and antibiotics to achieve the differentiated phenotype, as described in detail previously.1, 12
The AEC2 cells were seeded onto 24-well culture plates (105 cells per well) for ELISA studies, 6-well plates (2.5 × 105 cells per well) for PCR and FCM experiments, or 100-mm culture dishes (106 cells per dish) for western blot studies. Before treatment, the serum was replaced with only DMEM to make the cells serum starved for 12 h.
Flow Cytometric Analysis of Apoptosis
After the AEC2 cells were treated for 24 h, the cells were harvested and suspended in the binding buffer supplied in the Annexin V-FITC/Propidium iodide (PI) Apoptosis Detection Kit (eBioscience, San Diego, CA, USA). The cell suspensions (5 × 105–1 × 106 cells per ml) were labeled with FITC-conjugated Annexin V and PI and analyzed by flow cytometry. The percentage of cells in Q2 and Q4 were considered the apoptosis percentage in each experiment. The percentage of apoptosis in the control group was set to 1 for each experiment, and the other groups were analyzed relative to relevant control group.
Cytokine and Chemokine Assays
After the AEC2 cells were stimulated for 24 h, the culture supernatants were collected. The levels of multiple cytokines and chemokines were assayed using membranes from a Human Protein Cytokine Array Kit (Human Cytokine Antibody Array; RayBiotech, Norcross, GA, USA), according to the manufacturer’s instructions. Duplicate measurements for each treatment were obtained. In addition, the levels of IL-6, IL-8, TNF-α, and MCP-1 were measured using commercially available ELISA Kits (eBioscience), according to the manufacturer’s instructions. Each treatment was measured in triplicate.
Western Blot Analysis
Equal amounts of whole-cell extracts were electrophoresed on sodium dodecyl sulfate-polyacrylamide gels and then transferred onto polyvinylidenedifluoride membranes (Millipore, Bedford, MA, USA). Western blot analysis was performed using the Image J2x software (version2.1.4.7; National Institutes of Health, Bethesda, MD, USA).
Statistical Analysis
The data are expressed as the means±s.e.m. of independent experiments. Changes in IκB-α and p-IκB-α protein levels were analyzed using a two-way ANOVA (treatment and time). All other data were analyzed using a one-way ANOVA with a Bonferroni post-test for multiple comparisons. All of the statistical analyses were performed using GraphPad Prism (version 5.0; GraphPad Software, San Diego, CA, USA). A difference was considered significant at a P-value of ≤0.05 or less.
RESULTS
Purity and Viability of Primary Cultured AEC2 Cells
Monolayers of AEC2 cells were counted under light microscopy and photographed at × 200 magnification. Results showed that there was no significant difference cell density between plates, although little different seeding concentrations. After adherence and culture for 48 h non-adherent cells were washed away. With the immunohistochemical method, the staining-positive AECII cells could be counted under microscope and the mean purity was all above 90% (Figure 1a). Electron microscopy revealed that the cells had lamellar bodies and microvilli (Figure 1b).

Purity of primary cultured alveolar epithelial type 2 cells (AEC2 cells). (a) With the immunohistochemical method, the staining-positive AEC2 cells were counted under phase-contrast microscope and photographed at × 200 magnification. The mean purity was all above 90%. (b) Transmission electron microscope showed the lamellar bodies.
To determine viable cell percentage, cells were stained with a 0.25% solution of trypan blue before any treatment. The average starting percentage of each group showed no significant difference compare with the control group and was about 92.4±5% (P<0.05).
LPS Induces Caspase-Dependent Apoptosis of AEC2 Cells
To determine whether LPS induces apoptosis of AEC2 cells, the cells were stimulated with 1 or 10 μg/ml of LPS for 24 h. Using flow cytometry, we quantified LPS-induced apoptosis of AEC2 cells after the cells were stained with PI/Annexin V. Compared with the control group, LPS stimulation of the AEC2 cells triggered a low level of apoptosis. To determine if LPS-induced cell death of AEC2 cells was mediated via the caspase pathway, AEC2 cells were incubated with the pancaspase inhibitor Z-VAD (100 μM) for 1 h before LPS treatment (10 μg/ml). Pretreatment with Z-VAD blocked the proapoptotic effects of LPS in AEC2 cells (Figures 2a and b).

Lipopolysaccharide (LPS) induces apoptosis of alveolar epithelial type 2 cells (AEC2 cells) and the cleavage of caspase-3. (a) AEC2 cells were incubated with 1 or 10 μg/ml of LPS for 24 h. One group of the AEC2 cells was pretreated with Z-VAD and then treated with 10 μg/ml of LPS. Apoptosis of the AEC2 cells was evaluated by flow cytometry after the cells were stained with fluorescein isothiocyanate (FITC)-labeled Annexin V and propidium iodide (PI). (b) The graph represents the level of apoptosis relative to the values in the control group. (c) AEC2 cells were treated as in (a). The cytoplasmic extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with a caspase-3 antibody. The same extracts were immunoblotted with an antibody against β-actin, which was used as the control. A representative result of three independent experiments is shown. (d) The caspase-3 and cleaved caspase-3 bands from the experiments in (c) were normalized to β-actin and are expressed relative to the levels observed in the control group. The protein quantification was performed using densitometric analysis. The data are presented as the mean±s.e.m. of three independent experiments. *P<0.05; **P<0.01 compared with the control group; §§P<0.01 compared with the group that received Z-VAD before the administration of LPS.
To exclude the possibility that LPS-induced cell death of AEC2 cells was spontaneous, that is, not dependent on the activity of caspases, we measured the activity of caspase-3 after LPS treatment by western blot. Compared with the control group, LPS treatment resulted in activated caspase-3 cleavage; this activation was blocked by Z-VAD (Figures 2c and d).
RvD1 Protects AEC2 Cells Against LPS-induced Apoptosis
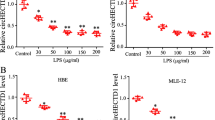
We evaluated the effects of RvD1 on apoptosis of LPS-stimulated AEC2 cells. AEC2 cells were incubated with a vehicle control or different concentrations of RvD1 (1, 10, or 100 ng/ml) for 30 min and then stimulated with 10 μg/ml of LPS for 24 h. Compared with the control group, LPS induced significant apoptosis of AEC2 cells. Pretreatment with RvD1 significantly reduced LPS-induced apoptosis of AEC2 cells (Figures 3a and b).

ResolvinD1 (RvD1) protects alveolar epithelial type 2 cells (AEC2 cells) against lipopolysaccharide (LPS)-induced apoptosis. (a) AEC2 cells were incubated with a vehicle control or different concentrations of RvD1 (1, 10, and 100 ng/ml) for 30 min before treatment with 10 μg/ml of LPS for 24 h. Flow cytometry analysis was performed as described previously. (b) The graph represents the level of apoptosis relative to the values in the control group. (c and d) The expression of caspase-3, Bax, and Bcl-2 was analyzed by western blot. A representative result of three independent experiments is shown. *P<0.05 compared with the control group; **P<0.01 compared with the control group; §P<0.05 compared with the LPS-treated group; and §§P<0.01 compared with the LPS-treated group. FITC, fluorescein isothiocyanate; PI, propidium iodide.
Apoptosis is accompanied by caspase-3 cleavage, the upregulation of Bax, and the downregulation of Bcl-2. To investigate whether RvD1-induced inhibition of apoptosis is regulated by caspase-3 and proteins in the Bcl-2 family, AEC2 cells were pretreated with 100 ng/ml of RvD1 for 30 min before treatment with 10 μg/ml of LPS for 24 h. RvD1 markedly inhibited caspase-3 cleavage, Bax upregulation, and Bcl-2 downregulation. In addition, RvD1 alone did not affect caspase-3 cleavage or the expression of Bax and Bcl-2 relative to the levels observed in the control group (Figures 3c and d).
RvD1 Activates the Phosphorylation of AKT in AEC2 Cells
The PI3K/AKT signaling pathway has an important role in cell survival. To investigate whether RvD1 is able to activate AKT phosphorylation in LPS-stimulated AEC2 cells, AEC2 cells were exposed to different concentrations of RvD1 (1, 10, and 100 ng/ml) for 30 min and then incubated with 10 μg/ml of LPS for 24 h. The RvD1-treated AEC2 cells exhibited a high amount of AKT phosphorylation, which was dependent on the concentration of RvD1 (Figures 4a and b). In addition, we examined whether RvD1 alone had effects on phosphorylation of AKT, and whether AEC2 cells were exposed to different concentration of RvD1 (1, 10, and 100 ng/ml) alone for 24 h. As a result, we found that RvD1 activate AKT phosphorylation again without the stimulation of LPS (Figures 4c and d).

ResolvinD1 (RvD1) activates the phosphorylation of AKT in alveolar epithelial type 2 cells (AEC2 cells). (a) AEC2 cells were treated as in Figure 2. Cytoplasmic extracts were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotted with an anti-phosphorylated AKT antibody. The same extracts were immunoblotted using an AKT antibody, which was used as the control. (b) The phosphorylated AKT (p-AKT) bands from the experiments in (a) were normalized to AKT and are expressed relative to the levels observed in the control group. The protein quantification was performed using densitometric analysis. (c) AEC2 cells were cultured with different concentrations of RvD1 (1, 10, and 100 ng/ml) for 24 h. Western blotS were carried out as described previously. A representative result of three independent experiments is shown. (d) The protein normalization and quantification were carried out as described previously. **P<0.01 compared with the control group; §§P<0.01 compared with the group that received 100 ng/ml of RvD1 before LPS treatment; and ##P<0.01 compared with the group that received 10 ng/ml of RvD1.
RvD1 Regulates Apoptosis by Inducing Phosphorylation of AKT in LPS-induced AEC2 Cells
To determine if RvD1-induced inhibition of LPS-induced apoptosis of AEC2 cells is associated with AKT phosphorylation, AEC2 cells were pretreated with wortmannin (200 nM, PI3K inhibitor), LY294002 (1 μM, PI3K inhibitor), PD98059 (50 μM, ERK inhibitor), or a vehicle control before the addition of 100 ng/ml of RvD1 and subsequent administration of 10 μg/ml of LPS. The PI3K inhibitors (wortmannin and LY294002) prevented the RvD1-induced protection against LPS-induced apoptosis of AEC2 cells (Figures 5a and b). In addition, the inhibition of AKT phosphorylation abrogated the reversal of RvD1-induced Bax upregulation in the LPS-induced AEC2 cells. However, no significant difference in the downregulation of Bcl-2 was observed (Figures 5c and d).

ResolvinD1 (RvD1) regulates apoptosis by inducing AKT phosphorylation in lipopolysaccharide (LPS)-stimulated alveolar epithelial type 2 cells (AEC2 cells). (a) All the experiments were carried out in the presence of RvD1 and LPS, and AEC2 cells were exposed to wortmannin (200 nM), LY294002 (1 μM), PD98059 (50 μM), or a vehicle control 1 h before RvD1 pretreatment and the subsequent administration of LPS. The cells were harvested and analyzed by flow cytometry as described previously. (b) The graph represents the level of apoptosis relative to that observed in the vehicle group. **P<0.01 compared with the vehicle group. (c) AEC2 cells were treated as in panel a. The levels of phosphorylated AKT (p-AKT), AKT, Bcl-2, and Bax were analyzed by western blot. A representative result of three independent experiments is shown. (d) The p-AKT bands from the experiments in c were normalized to AKT; the Bcl-2 and Bax bands were normalized to β-actin. All of the values are expressed relative to the vehicle group. **P<0.01 compared with the vehicle group. FITC, fluorescein isothiocyanate; PI, propidium iodide.
RvD1 Inhibits the LPS-induced Release of Chemokines and Cytokines in AEC2 Cells
To identify the anti-inflammatory effects of RvD1, a human cytokine antibody array was used to analyze the production of chemokines and cytokines in the conditioned medium collected from LPS-stimulated AEC2 cells in the presence or absence of RvD1. The levels of most of the chemokines and proinflammatory cytokines produced by the AEC2 cells, including TNF-α, RANTES, MCP-1, and IL-6, decreased as a result of RvD1 pretreatment (Figures 6a and b).

Inhibition of cytokine and chemokine production by resolvinD1 (RvD1) in lipopolysaccharide (LPS)-stimulated alveolar epithelial type 2 cells (AEC2 cells). (a) AEC2 cells were either untreated or pretreated with RvD1 for 30 min and then exposed to LPS for 24 h. The cytokines and chemokines in the conditioned medium were analyzed using a Human Protein Cytokine Array Kit. The upper line shows the antibody map of the membranes that were supplied in the kit. The lower line shows the representative result of two independent experiments. (b) The cytokines analyzed in the experiment in (a) were quantified using densitometric analysis according to the antibody map. The levels were normalized with background subtraction and are expressed relative to the values in the LPS-treated group. (c) AEC2 cells were exposed to different concentrations of RvD1 (1, 10, and 100 ng/ml) for 30 min and then to 1 μg/ml of LPS for 24 h. The levels of interleukin-6 (IL-6), tumor necrosis factor (TNF-α), IL-8 and monocyte chemoattractant protein-1 (MCP-1) in the conditioned medium were measured using commercially available ELISA Kits. The data are presented as the mean±s.e.m. of three independent experiments. *P<0.05 compared with the control group; **P<0.01 compared with the control group; §P<0.05 compared with the LPS-treated group; and §§P<0.01 compared with the LPS-treated group. G-CSF, granulocyte-colony stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; GRO, growth-regulated gene product.
To validate the findings obtained from the cytokine array experiment, we measured the levels of IL-6, TNF-α, IL-8, and MCP-1 in the conditioned medium (Figure 6c). The AEC2 cells were incubated in the presence or absence of different concentrations of RvD1 (1, 10, and 100 ng/ml) for 30 min and then stimulated with 100 ng/ml of LPS for 24 h. Consistent with the array results, LPS stimulation significantly induced the production of proinflammatory cytokines (IL-6, TNF-α) and chemokines (IL-8, MCP-1) in AEC2 cells. This production was significantly inhibited upon pretreatment with RvD1 (Figure 6c).
RvD1 Exhibits Anti-Inflammatory Effects Through the Inhibition of IκB-α Phosphorylation and Degradation
The LPS-induced production of cytokines and chemokines is associated with the activation of Toll-like receptor 4 and downstream signaling events, such as the phosphorylation and degradation of IκB-α.15 In the current study, we observed significant phosphorylation and degradation of IκB-α in LPS-stimulated AEC2 cells. To determine whether RvD1 exerts anti-inflammatory effects through the regulation of the phosphorylation and degradation of IκB-α, we examined the effects of RvD1 on AEC2 cells at different time points. Western blot analysis revealed that LPS-induced phosphorylation and degradation of IκB-α was significantly prevented when the cells were pretreated with 100 ng/ml of RvD1 (Figures 7a and b).

ResolvinD1 (RvD1)-induced inhibition of lipopolysaccharide (LPS)-induced IκB-α phosphorylation and degradation in alveolar epithelial type 2 cells (AEC2 cells). (a) AEC2 cells were either untreated or pretreated with 100 ng/ml of RvD1 and then exposed to 1 μg/ml LPS for the indicated times. The protein expression levels of IκB-α and phosphorylated IκB-α (p-IκB-α) were analyzed by western blot. A representative result of three independent experiments is shown. (b) The protein levels were quantified using densitometric analysis and are expressed relative to the values in the control group at the 0 time point. *P<0.05 compared with the LPS-treated group; **P<0.01 compared with the LPS-treated group.
Inhibition of NF-κB Activity Does Not Alter LPS-Induced Apoptosis of AEC2 Cells
Given the proapoptotic activity of NF-κB, we investigated the possibility that RvD1-induced inhibition of LPS-induced apoptosis of AEC2 cells is the result of the inhibition of NF-κB. AEC2 cells were incubated with a vehicle control, an irreversible inhibitor of IκB degradation (BAY-11-7082) or RvD1 and then treated with LPS. The inhibition of LPS-induced IκB degradation by BAY-11-7082 did not affect LPS-induced apoptosis of AEC2 cells (Figures 8a and b). The levels of IκB were evaluated by western blot (Figures 8c and d).

Inhibition of IκB degradation by BAY-11-7082 does not alter lipopolysaccharide (LPS)-induced apoptosis of alveolar epithelial type 2 cells (AEC2 cells). (a) AEC2 cells were pretreated with a control, RvD1 (100 ng/ml) or BAY-11-7082 (12 μM) for 30 min and then exposed to 10 μg/ml of LPS for 24 h. The levels of apoptosis were analyzed by flow cytometry. A set of representative results from three independent experiments is shown. (b) The graph represents the level of apoptosis relative to the values in the control group. (c) AEC2 cells were subjected to the same pretreatment as in (a) and then exposed to 10 μg/ml of LPS for 30 min. The level of IκB-α was analyzed by western blot. (d) The quantification of IκB-α level was performed using densitometric analysis and is expressed relative to the levels in the control group. *P<0.05 compared with the control group. **P<0.01 compared with the control group; §P<0.05 compared with the LPS-treated group; and §§P<0.01 compared with the LPS-treated group. FITC, fluorescein isothiocyanate; PI, propidium iodide.
DISCUSSION
As a part of the inflammation resolution process, the reduction of inflammatory mediators and the restoration of epithelial integrity are important steps in normal wound repair.3 In acute lung injury, alveolar epithelial cells undergo extensive apoptosis after the intratracheal administration of LPS into the lungs of the mice.4 AEC2 cells have been shown to release high levels of cytokines and, in particular, chemokines upon stimulation with different concentrations of LPS.1 Our data confirm the previously observed inflammatory response in AEC2 cells following LPS treatment. However, in contrast to previous studies,16, 17 we observed a low but significant level of apoptosis in AEC2 cells that were treated with LPS (1 μg/ml). This finding may be the result of the use of different cell types. Other groups used a cell line composed of type 2 pneumocytes (A549), while we chose to use primary human type 2 alveolar epithelial cells in our studies. Nevertheless, A549 cells exposed to LPS have been shown to exhibit apoptotic features based on an increase in the number of Annexin V-positive cells after LPS stimulation, which is similar to our results.17 Thus, the LPS stimulation of AEC2 cells results in the induction of an inflammatory response and apoptosis. In addition, we demonstrated that RvD1 inhibits LPS-induced apoptosis and the inflammatory response in AEC2 cells. These outcomes are regulated through the PI3K-AKT signaling pathway and the phosphorylation and degradation of IκB-α, respectively.
Apoptosis, which is a type of programmed cell death, regulates the homeostasis of lung tissue and host defense.18, 19 The survival and recovery of epithelial cells have important roles in normal lung tissue repair. Extensive evidence demonstrates that RvD1 and resolvin E1 exhibit anti-inflammatory and proresolution activities in acute lung injury.7, 8, 20 Animals treated with aspirin induced RvD1 and exhibited improved epithelial and endothelial barrier integrity.20 The protein flux in the bronchoalveolar lavage fluid in an animal model of an acute lung injury can be reduced by the induction of RvD1.8 In the present study, LPS-induced apoptosis of primary human alveolar epithelial cells was reduced by RvD1, which is consistent with RvD1-induced protection against epithelial barrier disruption and protein flux in vivo.
The PI3K-AKT signaling pathway regulates apoptosis, transcription, and proliferation.21, 22 Activated AKT protects murine alveolar epithelial cells from oxidant-induced injury.23 Recombinant mouse osteopontin induces the proliferation of human bronchial smooth muscle cells through the activation of the PI3K/AKT signaling pathway.24 The members of the Bcl-2 family of proteins, which are divided into the antiapoptotic Bcl-2 family and the proapoptotic Bax and BH3-only proteins,25 are important regulators of mitochondrial cytochrome c release. The expression levels of Bax and Bcl-2 are upregulated in alveolar epithelial cells of patients with diffuse alveolar damage.26, 27 The expression of the Bcl-2 protein family is regulated by multiple mechanisms, including the activation of the serine-threonine kinase AKT.28 The ability of IL-6 to protect against acute lung injury in vivo and to protect cells from oxidant-mediated death in vitro is partly the result of PI3K-AKT-mediated Bax phosphorylation.29 RvE1 has been shown to enhance the phagocytosis of human macrophages via the PI3K-AKT pathway.30 In addition, RvD1 and other Rvs can enhance macrophage phagocytosis and neutrophil apoptosis.11, 31, 32 Another DHA-derived mediator NPD1 also counter-regulates oxidative stress-triggered apoptosis in retinal pigment epithelium via upregulating the antiapoptotic proteins Bcl-2 and Bcl-x (L) and decreasing proapoptotic Bax and Bad expression.33 In this study, we observed that the effects of RvD1 on the LPS-induced apoptosis of AEC2 cells are largely mediated through the activation of the PI3K-AKT signaling pathway, and the inhibition of the PI3K-AKT pathway can block the RvD1-mediated expression of Bax.
It may be seem contrary to previous studies that RvD1 can increase apoptosis of T cells and neutrophils while our results show the apoptosis of AEC2 cells inhibited by RvD1. Complying with our results, studies have revealed that AT-RvD1 treatment after injury results in decreased hyperoxia-associated apoptotic markers in lung tissue34 and RvD1 attenuated ER stress-induced apoptosis and also decreased caspase-3 activity in human hepatoma cells.35 These effects may suggest that cells such as AEC2 cells, a kind of resident cells in lung tissue, in fact, may not be of the same function as those inflammatory cells such as neutrophils during injury.
In addition, previous research demonstrated that both suppressions of ALX/FPR2 and GPR32 did not significantly restore tunicamyin-induced apoptosis.35 We had tested the receptors of RvD1, ALX/FPR2, and GPR32 in the process and the RNA and protein expressions show significant increase even in LPS group. Thus, we concluded that the antiapoptotic effect of RvD1 in AEC2 cells might be an indirect effect. There could be several signaling pathway involved in the antiapoptotic mechanism. It was believed that RvD1 can reduce ER stress-induced apoptosis through JNK pathway in HepG2 cells35 as well as rescue macrophages from oxidative stress-induced apoptosis through PKA-mediated repression of NOX activation and upregulation of antiapoptotic protein expression.32 Some researchers even suspected that CCR5 involved in apoptosis and others indicated it was related with PMNs apoptotic effect.36, 37 IκB also increases the PMNs apoptosis but our results indicated that this pathway did not work in AEC2 cells.
In addition to apoptosis, we examined the anti-inflammatory effects of RvD1 in LPS-stimulated AEC2 cells. AEC2 cells are known to release a variety of mediators upon stimulation with proinflammatory agents.38, 39 Moreover, the alveolar epithelium is a rich source of chemokines2 that are able to recruit polymorphonuclear leukocytes to the local inflammatory site and may ultimately regulate the prolonged and unsuccessful resolution of inflammation.40, 41 Thus, the inhibition of chemokine production by epithelial cells serves as a key mechanism in the resolution of pulmonary inflammation. In the present study, we found that RvD1 markedly reduced LPS-induced production of cytokines and chemokines in AEC2 cells. This finding is consistent with the anti-inflammatory effects of another type of SPM (aspirin-triggered lipoxin A4) used in our previous study.42 However, in macrophages, 17R-RvD1 and RvD1 regulates LPS-induced primary human macrophage production of IL-7, IL-12p70, GM-CSF, IL-8, CCL2, and MIP-1α without reducing that of IL-6 and IL-10.43
It is well known that the activation of the transcription factor NF-κB regulates numerous genes that function in innate and adaptive immunity and the inflammatory response.44 There is extensive evidence that SPMs exhibit their anti-inflammatory effects through the inhibition of the NF-κB pathway.42, 45 IκBs are major regulators of NF-κB activity and are involved in the feedback inhibition of NF-κB. RvD1 markedly decreased airway eosinophilia and mucus metaplasia during allergic airway inflammation, in part, by decreasing IL-5 and IκBα degradation.46 In the present study, we focused on IκB phosphorylation and degradation and found that these processes are associated with the LPS-induced inflammatory response in AEC2 cells, which was attenuated upon pretreatment of the cells with RvD1. Considering the complex role of the pro- and antiapoptotic effects of NF-κB,47 we determined that IκB degradation did not contribute to LPS-induced apoptosis of AEC2 cells. Thus, we excluded the possibility that the RvD1-induced inhibition of LPS-induced apoptosis of AEC2 cells is due to the inhibition of IκB degradation.
In addition, MAPKs are also key signaling molecules that regulate the LPS/TLR4-mediated inflammatory process.48 Some existing experiments showed that G-protein coupled with receptors such as CCR5 would module inflammation and involve in cells apoptosis by MAPK- or PI3K-dependent signal pathway49 and lipoxin A4, resolvin E1, and protectin D1 can upregulate CCR5 in late apoptotic polymorphonuclear cells as terminators of chemokine signaling during the resolution of inflammation.36 However, a previous study showed that there was not an increase in phosphorylation of p38 MAPK and ERK1/2 following LPS stimulation in primary cultured human AEC2 cells. Moreover, the researchers discussed the involvement of the NF-κB transcription factor.1 Thus, we chose not to evaluate the MAPK signaling molecules. Although apoptosis and anti-inflammation can interact with each other in some experiments, the two physiology processes resulted from distinct pathways. It may be of significance to examine whether CCR5-PI3K-dependent signaling could be actived on primary cultured human AEC2 cells and regulate the anti-inflammation and apoptosis of AEC2 cells.
In summary, our study demonstrates that LPS induces the apoptosis of primary human epithelial cells in a caspase-dependent manner. In addition, apoptosis can be inhibited by RvD1 through the activation of the PI3K-AKT pathway. Moreover, RvD1 reduces the release of proinflammatory cytokines from LPS-stimulated AEC2 cells in a concentration-dependent manner. A possible mechanism for this anti-inflammatory effect involves the suppression of IκB phosphorylation and degradation. Because apoptosis and the inflammatory response in epithelial cells contribute to the pathogenesis of ALI/ARDS, pulmonary fibrosis and COPD, RvD1 may be a novel therapeutic agent for the treatment of these pulmonary disorders.
References
Thorley AJ, Ford PA, Giembycz MA et al. Differential regulation of cytokine release and leukocyte migration by lipopolysaccharide-stimulated primary human lung alveolar type II epithelial cells and macrophages. J Immunol 2007;178:463–473.
Witherden IR, Vanden BE, Goldstraw P et al. Primary human alveolar type II epithelial cell chemokine release: effects of cigarette smoke and neutrophil elastase. Am J Respir Cell Mol Biol 2004;30:500–509.
Xie W, Wang H, Wang L et al. Resolvin D1 reduces deterioration of tight junction proteins by upregulating HO-1 in LPS-induced mice. Lab Invest 2013;93:991–1000.
Kitamura Y, Hashimoto S, Mizuta N et al. Fas/FasL-dependent apoptosis of alveolar cells after lipopolysaccharide-induced lung injury in mice. Am J Respir Crit Care Med 2001;163:762–769.
de Souza PM, Lindsay MA . Apoptosis as a therapeutic target for the treatment of lung disease. Curr Opin Pharmacol 2005;5:232–237.
Eickmeier O, Seki H, Haworth O et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol 2013;6:256–266.
Seki H, Fukunaga K, Arita M et al. The anti-inflammatory and proresolving mediator resolvin E1 protects mice from bacterial pneumonia and acute lung injury. J Immunol 2010;184:836–843.
Wang B, Gong X, Wan JY et al. Resolvin D1 protects mice from LPS-induced acute lung injury. Pulm Pharmacol Ther 2011;24:434–441.
Keyes KT, Ye Y, Lin Y et al. Resolvin E1 protects the rat heart against reperfusion injury. Am J Physiol Heart Circ Physiol 2010;299:H153–H164.
Vasconcelos DP, Costa M, Amaral IF et al. Development of an immunomodulatory biomaterial: using resolvin D1 to modulate inflammation. Biomaterials 2015;53:566–573.
Krishnamoorthy S, Recchiuti A, Chiang N et al. Resolvin D1 binds human phagocytes with evidence for proresolving receptors. Proc Natl Acad Sci USA 2010;107:1660–1665.
Wang J, Oberley-Deegan R, Wang S et al. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza A infection. J Immunol 2009;182:1296–1304.
Fang X, Song Y, Hirsch J et al. Contribution of CFTR to apical-basolateral fluid transport in cultured human alveolar epithelial type II cells. Am J Physiol Lung Cell Mol Physiol 2006;290:L242–L249.
Ito Y, Correll K, Schiel JA et al. Lung fibroblasts accelerate wound closure in human alveolar epithelial cells through hepatocyte growth factor/c-Met signaling. Am J Physiol Lung Cell Mol Physiol 2014;307:L94–L105.
Christman JW, Sadikot RT, Blackwell TS . The role of nuclear factor-kappa B in pulmonary diseases. Chest 2000;117:1482–1487.
MacRedmond R, Singhera GK, Dorscheid DR . Erythropoietin inhibits respiratory epithelial cell apoptosis in a model of acute lung injury. Eur Respir J 2009;33:1403–1414.
Tang PS, Tsang ME, Lodyga M et al. Lipopolysaccharide accelerates caspase-independent but cathepsin B-dependent death of human lung epithelial cells. J Cell Physiol 2006;209:457–467.
Kuwano K . Epithelial cell apoptosis and lung remodeling. Cell Mol Immunol 2007;4:419–429.
Matute-Bello G, Martin TR . Science review: apoptosis in acute lung injury. Crit Care 2003;7:355–358.
Eickmeier O, Seki H, Haworth O et al. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol 2013;6:256–266.
Chang F, Lee JT, Navolanic PM et al. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia 2003;17:590–603.
Franke TF, Hornik CP, Segev L et al. PI3K/Akt and apoptosis: size matters. Oncogene 2003;22:8983–8998.
Sturrock A, Seedahmed E, Mir-Kasimov M et al. GM-CSF provides autocrine protection for murine alveolar epithelial cells from oxidant-induced mitochondrial injury. Am J Physiol Lung Cell Mol Physiol 2012;302:L343–L351.
Simoes DC, Xanthou G, Petrochilou K et al. Osteopontin deficiency protects against airway remodeling and hyperresponsiveness in chronic asthma. Am J Respir Crit Care Med 2009;179:894–902.
Cory S, Adams JM . The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002;2:647–656.
Guinee DJ, Brambilla E, Fleming M et al. The potential role of BAX and BCL-2 expression in diffuse alveolar damage. Am J Pathol 1997;151:999–1007.
Guinee DJ, Fleming M, Hayashi T et al. Association of p53 and WAF1 expression with apoptosis in diffuse alveolar damage. Am J Pathol 1996;149:531–538.
Datta SR, Dudek H, Tao X et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997;91:231–241.
Kolliputi N, Waxman AB . IL-6 cytoprotection in hyperoxic acute lung injury occurs via PI3K/Akt-mediated Bax phosphorylation. Am J Physiol Lung Cell Mol Physiol 2009;297:L6–L16.
Ohira T, Arita M, Omori K et al. Resolvin E1 receptor activation signals phosphorylation and phagocytosis. J Biol Chem 2010;285:3451–3461.
Lee HN, Kundu JK, Cha YN et al. Resolvin D1 stimulates efferocytosis through p50/p50-mediated suppression of tumor necrosis factor-alpha expression. J Cell Sci 2013;126:4037–4047.
Lee HN, Surh YJ . Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem Pharmacol 2013;86:759–769.
Mukherjee PK, Marcheselli VL, Serhan CN et al. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci USA 2004;101:8491–8496.
Cox RJ, Phillips O, Fukumoto J et al. Enhanced resolution of hyperoxic acute lung injury as a result of aspirin triggered resolvin D1 treatment. Am J Respir Cell Mol Biol 2015;53:422–435.
Jung TW, Hwang HJ, Hong HC et al. Resolvin D1 reduces ER stress-induced apoptosis and triglyceride accumulation through JNK pathway in HepG2 cells. Mol Cell Endocrinol 2014;391:30–40.
Ariel A, Fredman G, Sun YP et al. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol 2006;7:1209–1216.
Vassiliou EK, Kesler OM, Tadros JH et al. Bone marrow-derived dendritic cells generated in the presence of resolvin E1 induce apoptosis of activated CD4+ T cells. J Immunol 2008;181:4534–4544.
Koyama S, Sato E, Nomura H et al. The potential of various lipopolysaccharides to release IL-8 and G-CSF. Am J Physiol Lung Cell Mol Physiol 2000;278:L658–L666.
McRitchie DI, Isowa N, Edelson JD et al. Production of tumour necrosis factor alpha by primary cultured rat alveolar epithelial cells. Cytokine 2000;12:644–654.
Serhan CN, Brain SD, Buckley CD et al. Resolution of inflammation: state of the art, definitions and terms. FASEB J 2007;21:325–332.
Serhan CN, Savill J . Resolution of inflammation: the beginning programs the end. Nat Immunol 2005;6:1191–1197.
Wang YP, Wu Y, Li LY et al. Aspirin-triggered lipoxin A4 attenuates LPS-induced pro-inflammatory responses by inhibiting activation of NF-kappaB and MAPKs in BV-2 microglial cells. J Neuroinflammation 2011;8:95.
Palmer CD, Mancuso CJ, Weiss JP et al. 17(R-Resolvin D1 differentially regulates TLR4-mediated responses of primary human macrophages to purified LPS and live E. coli. J Leukoc Biol 2011;90:459–470.
Vallabhapurapu S, Karin M . Regulation and function of NF-kappaB transcription factors in the immune system. Annu Rev Immunol 2009;27:693–733.
Herrera BS, Ohira T, Gao L et al. An endogenous regulator of inflammation, resolvin E1, modulates osteoclast differentiation and bone resorption. Br J Pharmacol 2008;155:1214–1223.
Rogerio AP, Haworth O, Croze R et al. Resolvin D1 and aspirin-triggered resolvin D1 promote resolution of allergic airways responses. J Immunol 2012;189:1983–1991.
Perkins ND . The diverse and complex roles of NF-kappaB subunits in cancer. Nat Rev Cancer 2012;12:121–132.
Lu YC, Yeh WC, Ohashi PS . LPS/TLR4 signal transduction pathway. Cytokine 2008;42:145–151.
Lefkowitz RJ, Shenoy SK . Transduction of receptor signals by beta-arrestins. Science 2005;308:512–517.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (81070060 and 81370112).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
In this study the authors examine the mechanism of resolvin D1 (RvD1) in the amelioration of inflammatory lung disease. They show that RvD1 reduces apoptosis by inducing the phosphorylation of the kinase AKT and attenuates pro-inflammatory responses by suppressing the degradation and phosphorylation of the transcription factor inhibitor IκB-α.
Rights and permissions
About this article

Cite this article
Xie, W., Wang, H., Liu, Q. et al. ResolvinD1 reduces apoptosis and inflammation in primary human alveolar epithelial type 2 cells. Lab Invest 96, 526–536 (2016). https://doi.org/10.1038/labinvest.2016.31
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2016.31
This article is cited by
-
Functions of resolvin D1-ALX/FPR2 receptor interaction in the hemoglobin-induced microglial inflammatory response and neuronal injury
Journal of Neuroinflammation (2020)
-
Lipoxin A4 ameliorates lipopolysaccharide-induced lung injury through stimulating epithelial proliferation, reducing epithelial cell apoptosis and inhibits epithelial–mesenchymal transition
Respiratory Research (2019)
-
JAK2/STAT1-mediated HMGB1 translocation increases inflammation and cell death in a ventilator-induced lung injury model
Laboratory Investigation (2019)
-
Tetraspanin 1 inhibits TNFα-induced apoptosis via NF-κB signaling pathway in alveolar epithelial cells
Inflammation Research (2018)
