
Abstract
Coeliac disease (CD) is a malabsorptive enteropathy resulting from intolerance to gluten. Environmental factors and the microbiota are suggested to have critical roles in the onset of CD. The CD71 IgA receptor on epithelial cells is responsible for abnormal retrotranscytosis of IgA-gluten peptide complexes from the intestinal lumen into the lamina propria, inducing intestinal inflammation. However, understanding the role of gluten in the CD physiopathology has been hindered by the absence of relevant animal models. Here, we generated a mouse model for CD to study the factors controlling its pathogenesis as well as to investigate the influence of oral delivery of probiotics on disease development. Gluten sensitivity was established by feeding three generations of BALB/c mice a gluten-free diet (G−) followed by gluten challenge (G+) for 30 days. The G+ mice developed villous atrophy, crypt hyperplasia and infiltration of T cells and macrophages in the small intestine. Inflammation was associated with an overexpression of CD71 on the apical side of enterocytes and an increase of plasma cells producing IgA, which colocalised with the CD71. Moreover, IgA colocalised with the transglutaminase 2 (TG2), the production of which was increased in the lamina propria of G+ mice. These mice displayed increased production of cyclooxygenase-2 (COX-2), pro-inflammatory cytokines and IL-15, as well as anti-gliadin and anti-TG2 autoantibodies. The commensal flora-isolated presumptive probiotic Saccharomyces boulardii KK1 strain hydrolysed the 28-kDa α-gliadin fraction, and its oral delivery in G+ mice improved enteropathy development in association with decrease of epithelial cell CD71 expression and local cytokine production. In conclusion, the G+ BALB/c mouse represents a new mouse model for human CD based on histopathological features and expression of common biomarkers. The selected probiotic treatment reversing disease development will allow the study of the role of probiotics as a new therapeutic approach of CD.
Similar content being viewed by others


Main
A diet with gluten triggers the development of an enteropathy called coeliac disease (CD) in genetically susceptible individuals.1 CD is characterised by small intestinal mucosal injury, tissue remodelling and nutrient malabsorption.1 The gold standard for CD diagnosis includes the observation of villous blunting, crypt hyperplasia and intraepithelial lymphocytosis in intestinal biopsies. Gluten elicits T-and B-lymphocyte responses, and the detection of serum IgA and IgG anti-gliadin antibodies (AGA) as well as antibodies against tissue transglutaminase 2 (TG2) are also used for CD diagnosis.1, 2
The mechanism by which gluten peptides are transported intact across the mucosal epithelium is now becoming clear. Intact gliadin 33–mer or p31-49 peptides complexed with IgA are retrotranscytosed via the transferrin receptor (CD71/TfR1), an IgA receptor, which is overexpressed on the apical side of intestine epithelial cells in CD patients.3, 4 However, the factor promoting this overexpression is still unknown. The retrotranscytosis of IgA-gliadin complexes may trigger both innate and adaptive immune responses, which initiate CD enteropathy.5 In particular, in the lamina propria, gliadin peptides activate tissue TG2, the coeliac autoantigen,6 which selectively deamidates the gluten protein resulting in CD4+ T-cell activation. These cells release mediators, such as interferon-γ (IFN-γ),7 tumour necrosis factor-α (TNF-α)7 and interleukin (IL)-10,8 which ultimately cause tissue damage and the production of autoantibodies. During the innate immune response in CD, the expression of IL-15 is upregulated in the intestinal mucosa, resulting in the activation of cytotoxicity directed against enterocytes.9 Moreover, CD patients present increased production of inflammatory mediators such as cyclooxygenase-2 (COX-2), which contributes to the healing of the coeliac mucosa.10
A large number of clinical, genetic and immunological studies have been conducted on CD patients, revealing the crucial role of gluten in CD pathogenesis. Indeed, CD remission is highly dependent on a strict gluten-free diet.1 However, no animal model that recapitulates the disease process in terms of the histological, serological and immunological abnormalities of CD is available. The aim of this study was to investigate the development of gluten-induced enteropathy in BALB/c mice bred for three generations on a highly controlled gluten-free diet (G− mice) to achieve gluten intolerance. Gluten intolerance was tested by serum AGA production and spleen lymphoproliferative responses to gliadin. We report here that the exposure of G− mice to gluten (G+ mice) induced the development of multiple features of CD, including villus atrophy, crypt hyperplasia, infiltration of intraepithelial lymphocytes (IEL) associated with innate immunity reactions and anti-TG2 and AGA antibody production. Colocalisation experiments suggested that gluten triggers CD-like enteropathy by a mechanism that involves CD71, TG2 and IgA overexpression and interaction. This study highlights that the biological environment of the intestinal lumen has a crucial role in gluten-induced enteropathy, as our preclinical study revealed that Saccharomyces boulardii KK1, a presumptive probiotic strain, prevented CD-like enteropathy.
MATERIALS AND METHODS
Animals
Male or female 10-week-old BALB/c mice were inbred for at least three generations on a diet of gluten-free food pellets (G− mice). Gluten sensitivity was induced by feeding the G− mice gluten-containing commercial food pellets for 30 days (G+ mice). The strategy for preparing the gluten-free diet was to replace the gluten-containing ingredients with rice and fish meal, while keeping a similar content of protein, fat, fibre and minerals (Nutripet Hellas, Greece). All mouse experiments were performed at an accredited animal facility and were approved by the Hellenic Republic Prefecture of Thessaloniki (General Management Veterinary Service, Department of Veterinary Apprehension of Medicine and Applications).
The G− mice were immunised by administration of 50 μg whole gliadin emulsified in 50 μl of Freund′s complete adjuvant (FCA, DIFCO, Detroit, MI, USA) into each rear footpad, and serum was collected by day 10 post immunisation.
The G− mice received 50 μl of saline solution containing 109 CFU of each of the bacterial strains or 108 CFU of the KK1 strain orally for 7 consecutive days, whereas the control mice received only 50 μl of pyrogen-free saline solution. Then, the treated mice were fed a gluten diet for 30 days, whereas the oral administration of microorganisms continued twice per week. One day after the last dose, the intestine was removed and paraffin embedded.
Preparation of Gliadin
Wheat flour (10 g) was resuspended in 10% (w/v) NaCl, stirred for 1 h and then centrifuged at 6000 g for 5 min.11 The pellet was resuspended in 10% (w/v) NaCl, and the extraction procedure was repeated once again. The residue was further extracted twice with 70% (v/v) ethanol and centrifuged as described above. The supernatant was collected and filtered. The filtrate was lyophilised, and gliadin was dissolved in 70% ethanol containing 0.33 mM HCl.
Bacterial Strains and Growth Conditions
The Lactobacillus paracasei DC205 and DC41212 and Saccharomyces boulardii KK113 strains were obtained from the collection of the Laboratory of Food Microbiology and Hygiene of Aristotle University of Thessaloniki-Greece. All lactobacilli were stored at −80°C in MRS broth with 25% glycerol, and KK1 cultures were maintained at 4°C in a YEGP agar slope.14, 15 Yeast or lactobacilli were harvested at 18–24 h of culturing and resuspended at the appropriate concentration (CFU/ml) according to the McFarland standard.
Fermentation of Wheat Flour Sourdough
Lactobacilli or the KK1 strain (109 CFU) isolated at 8–10 h of culturing was mixed with 1 g of wheat flour in 4 ml of saline and incubated at 30°C for 24 h under stirring conditions.16 Gliadin was then isolated as described above. Samples were then subjected to western blot analysis as described below using serum isolated from G+ mice as the source of AGA.
Cell Lysates and Western Blot Analysis
The small intestine was homogenised in lysis buffer (50 mM tris-HCl pH 8.0, 1% Triton X-100, 1 μg/ml protease inhibitors of antipain and chymostatin) and cell lysates were prepared via repeated freeze-thaw treatments. TG2 levels in the small intestine cell lysates (20 μg) or the molecular weight of different gliadin forms (6 μg) were assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then electrotransferred to PVDF membranes and subjected to western blot analysis using the polyclonal anti-TG2 antibody (Thermo Scientific, Illkirch, France) or primary antibodies from mouse serum, respectively.
ELISA for IgA Anti-TG Antibody Detection
Plates were coated with 10 μg/well guinea pig liver tissue TG (Sigma, St Louis, MO, USA) in BBS overnight at 4°C. After washing, a blocking solution (BBS, 2% BSA) was used for 2 h. Mouse sera were incubated in PBS, 0.05% Tween, 0.1% azide and 1% BSA overnight at 4°C. After washing, the biotinylated mAb anti-mouse IgA (Southern Biotech, Birmingham, AL, USA) was added for 1 h, followed by incubation with streptavidin coupled with alkaline-phosphatase (AP, Jackson, West Grove, PA, USA). The reaction was developed by adding the AP substrate (Sigma).
Histopathology
Pieces of the small intestine (jejunum) were initially fixed in 4% paraformaldehyde, embedded in paraffin and cut into 4-μm sections onto gelatin-coated slides.14 The sections were stained with hematoxylin and eosin and viewed under regular light microscopy. The villus height/crypt depth (V:C) ratios were obtained from linear measurements of five villus heights in each sample, divided by the corresponding crypt depths as described.17
Immunohistological Analysis of the Small Intestine
Immunohistochemistry on paraffin sections was performed using an anti-mouse IL-5 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or an anti-CD71 (ZYMED Laboratories, Invitrogen, Carlsbad, CA, USA).18 Horseradish peroxidase-conjugated goat anti-rabbit IgG (Sigma) was used as secondary antibody. Positive cells were determined using PBS containing 0.5 mg/ml 3,3-diaminobenzidine-tetrahydrochloride (Sigma) and 0.01% H2O2 as substrate. The number of IgA+ cells was determined by direct immunofluorescence using an anti-mouse IgA antibody (Sigma). Indirect immunofluorescence was used to identify the cells producing cytokines and inflammatory mediators by using specific antibodies against mouse IFN-γ, TNF-α, IL-10 (R&D Systems, Minneapolis, MN, USA), IL-12B, IL-15, COX-1 and COX-2 (Santa Cruz Biotechnology).14 The number of positive cells was counted in 10 randomly chosen fields, and an average for each mouse was considered for statistical analysis.
The pieces of small intestine were frozen in OCT (Labonord, Templemars, France), cut into 4-μm sections and stored at −20°C. Immunohistochemistry was performed using a primary antibody against CD3 (DakoCytomation, Carpinteria, CA, USA) or biotinylated antibodies against CD11b and CD71 (BD-Pharmingen, San Jose, CA, USA).3 Unlabelled antibody (CD3) staining was followed by incubation with anti-rabbit IgG biotinylated antibody as the secondary antibody (Southern Biotech) and then with streptavidin-HRP (Vector Laboratories, Burlingame, CA, USA). The B lymphocytes were detected using anti-CD19 FITC (BD-Pharmingen).
For colocalisation experiments, frozen sections were incubated successively with anti-mouse IgA-FITC, anti-CD71-biotin, or rabbit anti-TG2 followed by incubation with streptavidin-Alexa 568 or the secondary antibody coupled with Alexa 568. Tissue sections were mounted, and slides were analysed with a laser scanning confocal microscope (LSM 510, Zeiss).
Splenocyte Response to Gliadin
Splenocytes were cultured (2.5 × 106 cells/well) in 1 ml of RPMI-1640 complete medium19 in the presence or absence of 125 μg/ml of gliadin for 96 h at 37°C and 5% CO2 atmosphere. During the last 18 h, 1 μCi tritiated thymidine per well was added, and splenocyte proliferation was determined by tritiated thymidine incorporation using a β-counter.
Statistical Analysis
The results of cell quantification in the intestine sections are reported as the mean±s.e.m., and comparisons were performed using the nonparametric Mann–Whitney U test. When indicated, multiple comparisons were performed by one-way analysis of variance (ANOVA) followed by Tukey's test. Data were considered statistically significant with P<0.05.
RESULTS
Gluten Induces Coeliac-Like Disease Lesions with Intraepithelial T-Cell Infiltrates
The clinical features of CD involve villous atrophy and crypt hyperplasia associated with IEL infiltration.1, 5 A gluten-containing diet was given to G− mice for 30 days to induce villous atrophy and crypt hyperplasia (Figure 1a), whereas no abnormalities were seen in G− mice maintained on a gluten-free diet. This was confirmed by morphometric evaluation of villus height:crypt depth ratio, showing that this ratio is lower in G+ mice (P<0.001) (Figure 1b). Moreover, apical enterocytes and tight junctions were damaged in these mice (Figure 1c). Diarrhoea, bloating (ballooned stomach) as well hair loss (data not shown) were observed in 20% of G+ mice fed with the gluten diet for 3–6 months. G+ mice also exhibited a marked increase of CD3+ cells in the intestinal epithelium compared with G− mice (Figure 1d; P<0.05), indicating that these IEL may participate in the activation of cytotoxicity that leads to tissue injury in the small intestine of G+ mice. All these observations resembled the clinical manifestations of human CD and thus, this experimental CD model mimics the development of disease pathology in humans and represents a valuable biological system for study of local immune responses induced by gluten.

Gluten induces histopathological features of coeliac-like disease and infiltration of intraepithelial lymphocytes (IEL) in G− mice. (a) Haematoxylin-Eosin (H-E) staining of the jejunal region of G− mice fed a gluten-free diet (left) and G− mice fed a gluten diet for 30 days (G+ mice, right) (original magnification, × 200) (n=6). (b) Morphometric evaluation of villus height:crypt depth (V:C) ratio at H-E-stained longitudinal sections of the small intestine of G− and G+ mice ( × 100 magnification, n=6). (c) H-E staining showing enterocyte degeneration in apical part of villi and damage of tight junctions in G+ compared with G− mice (original magnification, × 200). (d) Immunostaining for the detection of CD3+ infiltrates in the epithelium (black arrows) and the lamina propria in frozen intestine sections of G− and G+ mice. The graph shows the corresponding number of positive cells per mm2 counted in 10 fields per mouse at × 200 magnification. (n=4). The graph represents the mean±s.e.m. **P<0.01 and ***P<0.001 using the nonparametric Mann–Whitney's U test. Bars=20 μm. LPL, lamina propria lymphocytes.
Gluten Induces IgA Production and Enterocyte CD71 Overexpression
The immune response in CD is initiated by the abnormal intestinal transport of gliadin peptides, recently shown to be mediated by the epithelial cell IgA receptor CD71.3 However, there is no evidence on the factor triggering the abnormal overexpression of CD71 and the retrotranscytosis of IgA-gliadin complexes. To test the role of gluten in the induction of CD71 on the enterocytes of G+ mice, we therefore examined the distribution of CD71 by immunohistochemistry and observed an overexpression of CD71 in the small intestine of G+ mice compared with G− mice (Figures 2a and c). In particular, in the G− mice, CD71 expression was confined to the basolateral pole of the villous epithelial cells (Figures 2a and c) as well as in both the basal and apical poles of crypt epithelial cells (data not shown). In contrast, in the G+ mice, CD71 was strongly expressed across the epithelial layer, including the flat surface epithelium (Figures 2a and c).

CD71 expression and IgA-producing cells are increased in the small intestine of G+ mice. (a) A representative microphotograph (original magnification, × 630) of small intestine immunohistochemical labelling showing the expression of CD71 at the basal or apical pole of villus epithelial cells in the G− compared with G+ mice (n=5). Bar=15 μm. (b) The histogram shows the number of IgA-producing cells in the small intestine of the G− mice after gluten-diet challenge or not. The graph represents the mean±s.e.m. of the number of positive cells counted at × 400 magnification (n=6, **P<0.01 by Mann–Whitney's U test). (c) Double immunofluorescence and colocalisation (white, see arrows) of IgA (green) and CD71 (magenta) were observed at the apical surface of the epithelium in the G+ mice (n=5; original magnification, × 630). Bar=10 μm.
Immunofluorescence and confocal microscopy analysis of the small intestine sections showed an increased number of IgA+ cells in the intestinal epithelium and the lamina propria of G+ mice compared with G− mice (Figures 2b and c; P<0.01). Confocal microscopy analysis demonstrated the colocalisation (white) of IgA (green) and CD71 (magenta) at both the apical and the basal poles of the epithelial cells (Figure 2c) of G+ mice. The above results demonstrate that the delivery of gluten-sensitised G− mice in a diet with gluten induces abnormal enterocyte apical overexpression of CD71, which is colocalised with IgA, suggesting that this IgA receptor may transport IgA-gliadin complexes from the lumen to the intestinal submucosa as observed in CD patients.
Gluten-Induced Enteropathy Is Associated with TG2 Overexpression
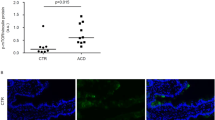
In CD patients, TG2 is a target for IgA antibodies20 so we next examined TG2 expression in our model. Figure 3 shows an increase of TG2 in the lamina propria of G+ mice as detected by immunofluorescence microscopy (Figures 3a and c; P<0.05). This was confirmed by western blot analysis of the intestinal cell lysates of these mice (Figure 3b), using a polyclonal anti-TG2 Ab. Confocal microscopy revealed an increased colocalisation (white) of TG2 (magenta) with IgA (green) in the lamina propria of the G+ mice compared with the G− mice (Figure 3c). In addition, increased levels of IgA anti-TG2 antibodies were observed in the serum of the G+ mice (Figure 3d; P<0.05). Together, these experiments suggested that gluten induces overexpression of TG2 in the small intestine of the G+ mice followed by increased IgA anti-TG2 antibody production.

Transglutaminase 2 (TG2) expression and colocalisation with IgA are increased in the small intestine of G+ mice. (a) Graph showing the number of TG2-producing cells as detected by immunofluorescence staining in paraffin sections of the small intestine of G− and G+ mice. TG2+ cells were counted and the graph represents the mean±s.e.m. *P<0.05 (Mann–Whitney's U test). (n=6). (b) A representative western blot analysis showed increased detection of TG2 (80 kDa) in the small intestine cell lysates of the G+ mice compared with the G− mice. Densitometric analysis of the bands using ImageJ software showed that the expression of TG2 was higher in the small intestine of the G+ mice (2.3:3.3) (n=3). (c) Double immunofluorescence staining of TG2-IgA in frozen sections of the small intestine. Colocalisation (white, see arrows) of TG2 (magenta) and IgA (green) was higher in the lamina propria of G+ mice (n=5). Bar=15 μm. (d) Detection of IgA anti-TG2 antibodies in the serum of G− and G+ mice by enzyme-linked immunosorbent assay (ELISA). Results are representative of three independent experiments. Data are presented in a box chart, where the boxes represent the 25th to 75th percentiles, the lines within the boxes represent the median, and the lines outside the boxes represent the 10th and 90th percentiles (n=8 G−, 11 G+, *P<0.05).
Gluten Elicits Specific Immune Responses to Gliadin
Gliadins are the primary pathogenic components of gluten, and they induce specific immune responses in CD patients that result in elevated serum levels of IgA and IgG AGA.2 We therefore addressed whether IgA and IgG AGA were induced in the G+ mice. As shown in Figures 4a and b, gluten delivery in the G− mice elicited a specific humoral immune response as demonstrated by the detection of specific IgA and IgG AGA in the serum. Although both isotypes reacted equally well with α- and γ-gliadin fractions, only IgA AGA strongly recognised the 62-kDa band of ω-gliadin, indicating a preferential activation by gluten of IgA responses in the gut. To further demonstrate that G− mice were sensitive to gliadin, we systemically immunised these mice with gliadin emulsified in FCA. Figure 4b shows that only the mice immunised with the FCA-gliadin emulsion contained IgG AGA in their serum, whereas no such antibodies were detectable in the mice immunised with FCA alone (Figure 4b). Furthermore, footpad FCA-gliadin s.c. administration did not elicit IgA AGA production in the serum of G− mice (data not shown). This indicates that, while systemic immunisation with gliadin preferentially induces IgG AGA responses, oral immunisation with gluten induces IgA AGA responses.

Specific immune response to gliadin in G+ mice. (a,b) Detection of serum anti-gliadin antibodies (AGA) by gliadin immunoblotting. Serum antibodies were revealed by biotinylated anti-IgA plus streptavidin horse radish peroxidase (HRP) (a) or anti-IgG conjugated to HRP (b). The α, γ and ω fractions of gliadin are indicated. Compared with mice immunised only with Freund′s complete adjuvant (FCA), the G− mice immunised with gliadin-FCA emulsion had detectable IgG AGA antibodies in the serum (b). Note the higher affinity of IgA antibodies against the ω-gliadin fraction. (c) Ex vivo proliferation capacity of G− and G+ splenocytes in response to gliadin. Splenocytes were cultured in the presence or absence of 125 μg/ml gliadin and splenocyte proliferation was determined by tritiated thymidine incorporation (3H-TdR). Note the increased 3H-TdR incorporation by the G+ splenocytes compared with that of the G− splenocytes. This experiment was performed twice with groups of three mice each time and the graph represents the mean±s.e.m. of these experiments (*P<0.05).
Native gliadin also increased the ex vivo proliferation of the splenocytes of G+ mice but not of G− mice (Figure 4c; P<0.05), showing that gliadin elicits specific systemic cellular immune responses in the G+ mice.
Together, these results demonstrate achievement of gluten intolerance in G− mice, as their delivery in gluten diet activate gliadin-specific immune responses similarly observed in CD patients.
Gluten Induces Adaptive and Innate Immunity Mediators in the Small Intestine
The retrotranscytosis of IgA-gliadin complexes to the lamina propria triggers an immune response that perpetuates intestinal inflammation.3 Indeed, an increased infiltration of IELs was observed in the small intestine of G+ mice as already shown in Figure 1d. Moreover, the G+ mice exhibited an increase of CD19+ cells in the lamina propria of the small intestine compared with G− mice (Figure 5a; P<0.05), indicating that B lymphocytes are recruited in the inflamed small intestine of G− mice. Furthermore, cytokines, which are important mediators driving Th1/Th2 immune responses, are involved in the mechanisms for tissue remodelling that lead to the villous atrophy and crypt hyperplasia in CD.20 Compared with those of the G− mice, the IFN-γ+ and IL-10+ cells in the lamina propria of the G+ mice were observed to be increased (Figure 5b; P<0.01). In contrast, there was no difference in the number of IL-5+ and IL-12+ cells between the G− and G+ mice (Figure 5b).

A gluten-enriched diet induces innate and adaptive immune responses by cell infiltration and cytokine production. (a) The increased infiltration of CD19+ B lymphocytes in the G+ mouse small intestine was detected by confocal microscopy (original magnification, × 400). Bar=10 μm. Quantification is shown on the bottom of the histograms (n=4). (b) The number of interferon (IFN)-γ- and interleukin (IL)-10-producing cells increased in the small intestine of G+ mice. Representative immunofluorescence microphotographs (original magnification, × 400) of IL-10+ cells (white arrows) are shown at the top. Bar=20 μm. The quantification of cytokine-producing cells is shown at the bottom of the histograms (n=6). (c) Graph showing the number of CD11b+ cells in cryosections of the small intestine of G+ compared with G− mice as detected by immunohistochemistry using an anti-mouse CD11b antibody (n=4). (d) Immunofluorescence staining for the detection of cells expressing pro-inflammatory factors in the small intestine of the G− and G+ mice. The number of IL-15+, tumour necrosis factor-α (TNF-α+), cyclooxygenase (COX)-1+ or COX-2+ cells was counted (n=6) and the graphs represent the mean±s.e.m. (*P<0.05 and **P<0.01 by Mann–Whitney's U test).
Innate responses also have a crucial role in CD pathogenesis, particularly with macrophage infiltration and pro-inflammatory cytokine production.1, 5 Increased accumulation of CD11b+ macrophages was observed in the lamina propria of the G+ mice (Figure 5c; P<0.05), and this accumulation was associated with an increased number of IL-15+, TNF-α+ and COX-2+ cells (Figure 5d; P<0.01). In contrast, there were no changes in the number of COX-1+ cells (Figure 5d). The data in G+ mice provide evidence for the activation of pro-inflammatory responses that are dependent on dietary exposure to gluten.
All the above data further support that this mouse model exhibits immunological characteristics that are similar to those observed in CD patients.
Saccharomyces Boulardii KK1 Strain, a Presumptive Probiotic, Improves Gluten-Induced Enteropathy
Because probiotics have been shown to hydrolyse gliadin in vitro,16 we examined whether presumptive probiotics with established immunoregulatory activity14, 15 have an effect in vivo in the gluten-induced mouse model for CD. The hydrolytic activity of L. paracasei DC412, DC205 or S. boulardii KK1 strains was first verified in vitro by preparing wheat flour fermentation in the presence of these strains. Among the tested strains only the KK1 strain exhibited specific proteolytic activity on the 28-kDa α-gliadin (Figure 6a), which is the most toxic fraction in CD.16 We next investigated whether changes in the microflora of the intestinal lumen by oral delivery of the KK1 strain modulate gluten enteropathy in G+ mice (Figure 6b). Oral administration of the KK1 strain resulted in a decrease in villous atrophy, V:C ratio (P<0.001) and cell accumulation in the lamina propria (Figures 6c and d), whereas delivery of the DC412 strain accelerated the appearance of clinical manifestations such as hair loss (data not shown). Moreover, administration of KK1 strain resulted in a decreased expression of CD71 in treated mice compared with G+ mice (Figure 6e). In particular, in the G+/KK1 mice, CD71 expression was confined to the basolateral pole of the villous epithelial cells (Figure 6e), whereas in the untreated G+ mice, CD71 was strongly expressed across the surface epithelium as shown previously in Figure 2a. The delivery of DC412 and DC205 strains had no effect on CD71 epithelial cell expression (data not shown). The KK1 strain treatment also resulted in a decreased number of TNF-α+ and IFN-γ+ cells and an increased number of COX-2+ cells in the lamina propria of the treated G+ mice compared with that in the untreated G+ mice (Figure 6f; P<0.05). These data suggest that the presumptive probiotic KK1 strain affects the environment of the intestinal lumen and prevents gluten-induced enteropathy.

The Saccharomyces boulardii, a presumptive probiotic, hydrolysed α-gliadin and improved the Coeliac disease (CD)-like enteropathy in G+ mice. (a) Specific in vitro hydrolysis of a 28-kDa α-gliadin fragment was accomplished by the KK1 strain. Gliadin was detected using serum isolated from G+ mice. Gliadin was also isolated from acidified dough without the microorganism inoculums that served as control. (b) Schematic presentation of the treatment with the Lactobacilli or the KK1 strain. The G− mice received the DC205 or DC412 or KK1 strains in phosphate-buffered saline (PBS) orally for 7 consecutive days, whereas the control mice received only PBS. Then, the treated mice were fed a gluten diet for 30 days, whereas the oral administration of microorganisms continued twice per week. (c) Villus height:crypt depth (V:C) ratio at H-E stained longitudinal sections of the small intestine of G− and G+ mice, treated or not with the different probiotic strains ( × 100 magnification, n=3). (d) Haematoxylin-Eosin staining of the jejunal region of G− and G+ mice treated or not with the S. boulardii KK1 strain (original magnification, × 400). Note the improvement of the intestinal architecture of the mice treated with the KK1 strain. (e) A representative microphotograph (original magnification, × 630) of small intestine immunohistochemical labelling showing the expression of CD71 at the basal pole of villus epithelial cells in the G− mice, an overexpression of CD71 across the epithelial surface in the G+ mice and a decreased expression of CD71 confined to the basolateral side of the epithelial cells in G+ mice treated with the KK1 strain (n=3). (f) The histogram shows the number of IgA-, cytokine- or transglutaminase 2 (TG2)-producing cells in the small intestine of the G+ mice treated with KK1, DC205 or DC412 compared with not-treated mice (n=3, *P<0.05 G+ to G−, #P<0.05 KK1 to G+) counted at × 400 magnification. Bars=15 μm. COX, cyclooxygenase; IFN, interferon; IL, interleukin; TNF, tumour necrosis factor.
DISCUSSION
CD is a common disorder that affects 0.5–1% of the population worldwide.2 Although several abnormalities have been described recently in CD patients, understanding the disease development has been hampered by the absence of an animal model. The key role of gluten in the pathogenesis of CD has been established through epidemiological studies and the observation that CD patients enter remission when they adhere to a gluten-free diet. Saharawi21 and Latin American22 populations experienced an increased prevalence of CD after changing their low-gluten diets to gluten-enriched diets. We thus hypothesised that the elimination of gluten from the diet would allow for newborns to acquire intolerance against gliadin-derived peptides that exacerbate the immune responses against orally introduced gliadin and trigger enteropathy. Our findings indicate gluten intolerance achievement by breeding three generations of mice fed a diet without gluten and challenged orally with gluten for 30 days. These mice exhibited specific systemic as well as local responses against gluten, including increased IgA or IgG serum AGA production and spleen lymphoproliferative responses. Increased AGA production as well as hair loss was observed in G+ mice 6 months post gluten diet (data not shown), a phenomenon indicating persistence in gluten intolerance. In addition, challenge of the G− mice with a gluten-containing diet increased anti-TG2 antibodies in the serum and affected the intestinal architecture, causing villus atrophy, crypt hyperplasia and IEL infiltration. The responses observed in this mouse model are the first to show that gluten induces the enteropathy characteristic of human CD.
Several pathogenic factors described in human CD were also observed in this gluten-induced coeliac-like disease model. First, the IgA1 receptor CD71 was found to be overexpressed at the apical and basal sides of enterocytes, as observed in CD patients.3 In the G+ mice, CD71 was colocalised with IgA at both apical and basal poles of the enterocytes, suggesting its potential role in IgA-gliadin complex retrotranscytosis. Indeed in CD patients, villous atrophy and increased epithelial renewal are associated with CD71 upregulation all over the surface epithelium, and this may be responsible for its missorting toward the apical pole of enterocytes. This is compatible with a role of CD71 in the endocytosis of IgA immune complexes from the apical cell surface.3 Second, we demonstrated an increased production of TG2, which was colocalised with IgA antibodies in the lamina propria of the G+ mice, as found in human pathology.20 This association suggests that TG2 may potentially convert gliadins into immunogenic peptides that form immune complexes with IgA. Moreover, IgA-gliadin complexes could be selectively retrotranscytosed with the help of tissue TG, as Katoh et al23 have shown that TG can crosslink gliadin peptides and promote the invagination step of endocytosis of CD71. Third, the intestinal alterations were associated by increased accumulation of CD11b+ cells, CD19+ B lymphocytes in the lamina propria and CD3+IEL, suggesting that gliadin peptides may elicit the activation of both innate and adaptive immunity mechanisms in the G+ mice. Fourth, an increased number of cytokine-producing cells were detected in the lamina propria of the G+ mice as observed in human CD.24 It is noteworthy that IL-15 has been described to upregulate intestinal CD71 expression in CD,9 which may explain the overexpression of both molecules in G+ mice. Altogether, this mouse model recapitulates the key features of the human disease and may be used to test new potential therapeutic interventions in CD.
Homoeostasis in the healthy intestine is achieved primarily by the interaction of prokaryotic cells comprising the lumen microflora with the eukaryotic cells forming the intestinal barrier.25 Intraluminal therapies of CD are based on the use of ancient variants or genetic modifications of wheat as well as the pretreatment of flour by means of sourdough fermentation using certain lactobacilli with subsequent proteolysis of gluten.16 Our results showed that oral administration of the S. boulardii KK1 strain in the G+ mice ameliorated the clinical manifestations of coeliac-like disease induced by gluten, decreased the local production of pro-inflammatory cytokines and downregulated the expression of CD71, which was confined to the basolateral pole of enterocytes. KK1 treatment of the G+ mice increased COX-2 production in the small intestine, which is potentially related to processes that maintain gut mucosa integrity.10 Indeed, previous studies have shown that COX synthesis is inhibited by indomethacin in gluten-sensitive DQ8 transgenic mice and resulted in intestinal lesions after simultaneous exposure to gluten and cholera toxin.26 Furthermore, our results revealed that, whereas the Th1-inducer L. paracasei DC412 strain accelerated coeliac-like disease in G+ mice, DC205 strain exhibits low immunoregulatory activity and had no effect. Interestingly, neither of these strains fermented wheat flour, in contrast to the gliadin hydrolytic effect of the protective KK1 strain. Moreover, another probiotic, L. lactis, increased Th1 responses and IFN-γ production in gluten-sensitive DQ8 transgenic mice without inducing intestinal lesions.27 Thus, the gluten-induced Th1 immune response alone may not be a sufficient abnormality to promote disease development, but may rather be an important component of a complex mechanism that enables immune cells to interact with the host gut to exert their autoreactivity to gluten. Finally, our data revealed a heterogeneous response of G+ mice to three different probiotic strain treatments, indicating the importance of the biological environment of the intestinal lumen in disease progression.
In conclusion, we present G+ BALB/c mice, a new animal model of gluten-induced enteropathy that closely resembles human CD. Using this model, we showed for the first time that gluten is responsible for the overexpression of the CD71, the receptor that allows IgA gliadin-complexes to translocate from the intestinal lumen into the lamina propria in active enteropathy. G+ BALB/c mice are an important tool for the study of the pathogenic mechanism of CD as well as the proof of concept of new treatments for CD. Oral delivery of the S. boulardii KK1 strain reduced epithelial cell CD71 expression and Th1 immune responses and ameliorated the histopathological features of gluten-induced enteropathy in the G+ mice, potentially indicating a new therapeutic approach for CD.
References
Kagnoff MF . Overview and pathogenesis of celiac disease. Gastroenterology 2005;128:S10–S18.
Craig D, Robins G, Howdle PD . Advances in celiac disease. Curr Opin Gastroenterol 2007;23:142–148.
Matysiak-Budnik T, Moura IC, Arcos-Fajardo M, et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J Exp Med 2008;205:143–154.
Moura IC, Centelles MN, Arcos-Fajardo M, et al. Identification of the transferrin receptor as a novel immunoglobulin (Ig)A1 receptor and its enhanced expression on mesangial cells in IgA nephropathy. J Exp Med 2001;194:417–425.
Maiuri L, Ciacci C, Ricciardelli I, et al. Association between innate response to gliadin and activation of pathogenic T cells in coeliac disease. Lancet 2003;362:30–37.
Dieterich W, Ehnis T, Bauer M, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med 1997;3:797–801.
Nilsen EM, Lundin KE, Krajci P, et al. Gluten specific, HLA-DQ restricted T cells from coeliac mucosa produce cytokines with Th1 or Th0 profile dominated by interferon gamma. Gut 1995;37:766–776.
Beckett CG, Dell′Olio D, Kontakou M, et al. Analysis of interleukin-4 and interleukin-10 and their association with the lymphocytic infiltrate in the small intestine of patients with coeliac disease. Gut 1996;39:818–823.
Maiuri L, Ciacci C, Auricchio S, et al. Interleukin 15 mediates epithelial changes in celiac disease. Gastroenterology 2000;119:996–1006.
Kainulainen H, Rantala I, Collin P, et al. Blisters in the small intestinal mucosa of coeliac patients contain T cells positive for cyclooxygenase 2. Gut 2002;50:84–89.
Maurano F, Siciliano RA, De Giulio B, et al. Intranasal administration of one alpha gliadin can downregulate the immune response to whole gliadin in mice. Scand J Immunol 2001;53:290–295.
Xanthopoulos V, Hatzikamari M, Adamidis T, et al. Heterogeneity of Lactobacillus plantarum isolates from feta cheese throughout ripening. J Appl Microbiol 2000;88:1056–1064.
Psomas E, Andrighetto C, Litopoulou-Tzanetaki E, et al. Some probiotic properties of yeast isolates from infant faeces and Feta cheese. Int J Food Microbiol 2001;69:125–133.
Kourelis A, Zinonos I, Kakagianni M, et al. Validation of the dorsal air pouch model to predict and examine immunostimulatory responses in the gut. J Appl Microbiol 2010;108:274–284.
Kourelis A, Kotzamanidis C, Litopoulou-Tzanetaki E, et al. Immunostimulatory activity of potential probiotic yeast strains in the dorsal air pouch system and the gut mucosa. J Appl Microbiol 2010;109:260–271.
De Angelis M, Rizzello CG, Fasano A, et al. VSL#3 probiotic preparation has the capacity to hydrolyze gliadin polypeptides responsible for Celiac Sprue. Biochim Biophys Acta 2006;1762:80–93.
Bethune MT, Borda JT, Ribka E, et al. A non-human primate model for gluten sensitivity. PLoS One 2008;3:e1614.
Avramidis N, Victoratos P, Yiangou M, et al. Adjuvant regulation of cytokine profile and antibody isotype of immune responses to Mycoplasma agalactiae in mice. Vet Microbiol 2002;88:325–338.
Yiangou M, Hadjipetrou-Kourounakis L . Effect of magnesium deficiency on interleukin production by Fisher rats: effect of interleukins on reduced in vitro lymphocyte responses to concanavalin A and lipopolysaccharide. Int Arch Allergy Appl Immunol 1989;89:217–221.
Korponay-Szabo IR, Halttunen T, Szalai Z, et al. In vivo targeting of intestinal and extraintestinal transglutaminase 2 by coeliac autoantibodies. Gut 2004;53:641–648.
Catassi C, Ratsch IM, Gandolfi L, et al. Why is coeliac disease endemic in the people of the Sahara? Lancet 1999;354:647–648.
Cataldo F, Montalto G . Celiac disease in the developing countries: a new and challenging public health problem. World J Gastroenterol 2007;13:2153–2159.
Katoh S, Midorikami J, Takasu S, et al. Involvement of membrane-bound transglutaminase in the invagination of transferrin into rat reticulocyte plasma membrane. Biol Pharm Bull 1994;17:1003–1007.
Jabri B, Sollid LM . Tissue-mediated control of immunopathology in coeliac disease. Nat Rev Immunol 2009;9:858–870.
Corthesy B, Gaskins HR, Mercenier A . Cross-talk between probiotic bacteria and the host immune system. J Nutr 2007;137:781S–790S.
D’Arienzo R, Stefanile R, Maurano F, et al. A deregulated immune response to gliadin causes a decreased villus height in DQ8 transgenic mice. Eur J Immunol 2009;39:3552–3561.
D’Arienzo R, Maurano F, Luongo D, et al. Adjuvant effect of Lactobacillus casei in a mouse model of gluten sensitivity. Immunol Lett 2008;119:78–83.
Acknowledgements
We thank Professor Nikos Tzanetakis, who kindly provided bacterial and yeast strains; Paschalis Apostolidis and Spyridoula Kazantzidou of ‘Nutripet Hellas’, who supplied the gluten-free food pellets; Samira Benadda for her technical assistance with confocal microscopy analysis; and Jean-Pierre Laigneau for his contribution to the preparation of figures. This work was supported by Agence National pour la Recherche.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict interest.
Additional information
Gluten-sensitive BALB/c mice represent a new animal model of gluten- induced enteropathy that closely resembles human celiac disease. In these animals, lesions are associated with IgA, CD71 and transglutaminase-2 interactions and local pro-inflammatory cytokine production. Probiotics improve this enteropathy, and suggest a new therapeutic approach in humans
Rights and permissions
About this article
Cite this article
Papista, C., Gerakopoulos, V., Kourelis, A. et al. Gluten induces coeliac-like disease in sensitised mice involving IgA, CD71 and transglutaminase 2 interactions that are prevented by probiotics. Lab Invest 92, 625–635 (2012). https://doi.org/10.1038/labinvest.2012.13
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2012.13
Keywords
This article is cited by
-
Gut-Ex-Vivo system as a model to study gluten response in celiac disease
Cell Death Discovery (2021)
-
Current and emerging therapies for coeliac disease
Nature Reviews Gastroenterology & Hepatology (2021)
-
Gut Microbiota and Celiac Disease
Digestive Diseases and Sciences (2016)
-
Novel players in coeliac disease pathogenesis: role of the gut microbiota
Nature Reviews Gastroenterology & Hepatology (2015)
-
Gluten exacerbates IgA nephropathy in humanized mice through gliadin–CD89 interaction
Kidney International (2015)
