
Abstract
Mutations in homeoprotein NKX2-5 are linked to human congenital heart disease, resulting in various cardiac anomalies, as well as in postnatal progressive conduction defects and occasional left ventricular dysfunction; yet the function of Nkx2-5 in the postnatal period is largely unexplored. In the heart, the majority of cardiomyocytes are believed to complete cell-cycle withdrawal shortly after birth, which is generally accompanied by a re-organization of chromatin structure shown in other tissues. We reasoned that the effects of the loss of Nkx2-5 in mice may be different after cell-cycle withdrawal compared with those of the perinatal loss of Nkx2-5, which results in rapid conduction and contraction defects within 4 days after the deletion of Nkx2-5 alleles (Circ Res. 2008;103:580). In this study, floxed-Nkx2-5 alleles were deleted using tamoxifen-inducible Cre transgene (Cre-ER) beginning at 2 weeks of age. The loss of Nkx2-5 beginning at 2 weeks of age resulted in conduction and contraction defects similar to the perinatal loss of Nkx2-5, however, with a substantially slower disease progression shown by 1° atrioventricular block at 6 weeks of age (4 weeks after tamoxifen injections) and heart enlargement after 12 weeks of age (10 weeks after tamoxifen injections). The phenotypes were accompanied by a slower and smaller degree of reduction of several critical Nkx2-5 downstream targets that were observed in mice with a perinatal loss of Nkx2-5. These results suggest that Nkx2-5 is necessary for proper conduction and contraction after 2 weeks of age, but with a substantially distinct level of necessity at 2 weeks of age compared with that in the perinatal period.
Similar content being viewed by others


Main
Nkx2-5 is a homeodomain-containing transcription factor, highly conserved among species and is one of the earliest cardiogenic markers1, 2 with expression continuing throughout adulthood.3, 4, 5 The targeted disruption of Nkx2-5 in mice causes embryonic lethality around embryonic (E) day 10.5, with retarded cardiac development.6, 7 Mice with ventricular-restricted Nkx2-5 deletion from E 8.0–8.5 survive but show progressive and advanced conduction defects, as well as left ventricular hypertrophy postnatally.8 Heterozygous mutations in human NKX2-5 cause various cardiac anomalies and postnatal progressive conduction defects and occasional left ventricular dysfunction.9, 10, 11, 12
The Nkx2-5 function has predominantly been studied in mouse embryos or stem cells when cardiomyocytes are proliferating.13, 14 To address the Nkx2-5 function at distinct stages of development, we recently generated tamoxifen-inducible Nkx2-5 knockout mice. Using mice with a perinatal loss of Nkx2-5, we have demonstrated that Nkx2-5 is critically important for cardiac conduction and contraction in the perinatal period as well. In these mice, atrioventricular (AV) block and heart enlargement appeared within 4 days after tamoxifen injection, leading to premature death due to cardiac dysfunction.15 Rapid disease progression was accompanied by the rapid decrease of several gene products important for both conduction and contraction, including the cardiac Na+ channel, Nav1.5(α), ryanodine receptor 2 and cardiac myosin light chain kinase (MLCK).15, 16
Our ongoing hypothesis is that Nkx2-5 actively regulates a critical set of genes in postnatal cardiomyocytes to maintain proper cardiac function. Less well understood is the function of Nkx2-5 from the time between perinatal development to adulthood. Recent studies demonstrate the formation of a population of new myocytes in the adult heart;17 although perhaps the majority of cardiomyocytes may be considered to lose their ability for proliferation (G1 cell-cycle arrest) and DNA synthesis during the first week after birth, and transit into cells with a highly specialized structure and function.18 Because G1 cell-cycle arrest is generally linked to morphological, metabolic and gene expression changes with a re-organization of the nuclear architecture and chromatin structure,19, 20 cardiomyocytes at G1 cell-cycle arrest may share these properties. We reasoned that this transition might change the cardiomyocyte responses to cardiac transcription factors, including Nkx2-5.
In this study, we deleted the Nkx2-5 gene beginning at 2 weeks of age, which resulted in a slow progressive conduction defect and heart enlargement, accompanied by a slow downregulation of critical genes that were important determinants for the phenotypes observed in the perinatal loss of Nkx2-5 mice.
MATERIALS AND METHODS
Inducible Nkx2-5 Knockout Mice
Floxed-Nkx2-5 mice8 were bred with transgenic mice carrying the Cre-ER gene under CMV promoter.21 Details of mouse generation have been described previously.15 To delete the floxed-Nkx2-5 gene, tamoxifen (1 mg/g body weight, i.p.) was injected into mice beginning at 2 weeks of age for 4 consecutive days. All animal care protocols fully conformed to the Association for the Assessment and Accreditation of Laboratory Animal Care, with approvals from the University of Florida Institutional Animal Care and Use Committee.
Telemetry ECG Recordings and Echocardiogram
Recording of telemetry ECG (Data Sciences International) was performed 3 days after implantation of wireless radiofrequency telemetry devices to avoid the effects of anesthesia on ECG. Telemetry data were analyzed using PowerLab software (ADInstruments) as described previously.22 An M-mode ultrasound imaging of the left ventricle of anesthetized mice (pentobarbital 60 mg/kg, i.p.) was obtained at the level of the papillary muscle from a parasternal window using an ultrasound biomicroscope with a single transducer with a frequency of 40 MHz (VisualSonics, Toronto, Canada).
Northern Blotting, Immunostaining and Histological Analyses
Northern blot analyses were carried out using the following probes: an Nkx2-5-coding probe, a PflMI-EcoRI fragment of mouse Nkx2-5 cDNA; atrial natriuretic factor (ANF) (330 bp PCR products for rat ANF, F, 5′-GGGGTAGGATTGACAGGATTGG-3′; R, 5′-CCGTGGTGCTGAAGTTTATTCG-3′); brain natriuretic peptide (BNP) (429 bp PCR products, F, 5′-TGGGGAGGCGAGACAAGGG-3′; R, 5′-TCTTCCTACAACAACTTCAGTGCG-3′); β-myosin heavy chain (βMHC) (oligonucleotide probe, 5′-GCTTTATTCTGCTTCCACCTAAAGGGCTGTTGCAAAGGCTCCAGGTCTGAGGGCTTC-3′); GAPDH (450 bp PCR products, F, 5′-TTCATTGACCTCAACTACAT-3′; R, 5′-GTGGCAGTGATGGCATGGAC-3′).
Immunostaining was carried out with the following primary antibodies: anti-Nkx2-5 pAb,5 and sarcomeric actinin (Sigma A7811). Fluorescent microscopic images were obtained using ZEISS Axiovert200M with or without Apotome. Histological analyses, including hematoxylin/eosin and Masson's trichrome staining, acetylcholine esterase staining in frozen tissue sections and whole-mount acetylcholine esterase staining, were carried out as described previously.15 Digitalized images from the AV node, the LV free wall in the longitudinal section at the level of nuclei and isolated ventricular myocytes were utilized for measurement using Image J software as described previously.23, 24, 25
Simultaneous Recording of Cardiac Contraction and Ca2+ Measurements
Rod-shaped cardiomyocytes with clear cross striations, staircase ends and surface membranes free from blebs isolated from approximately 24-week-old flox/flox or flox/flox/Cre mice were studied for simultaneous measurements of cardiomyocyte cell shortening and intracellular free calcium concentration, as described previously.16
Real-Time RT-PCR
Real-time RT-PCR was performed using inventoried Taqman Gene Expression Assays (Applied Biosystems): cardiac MLCK, Mm00615292; Scn5a, Mm00451971; RyR2, Mm0046587; ANF, Mm01255748; BNP, Mm00435304; βMHC (MHC7), Mm00600555. Data were normalized to β-actin expression (no. 4352933E). Duplicate experiments were averaged.
Statistical Analysis
Values among groups were compared using ANOVA and the Fisher PLSD post hoc test (StatView version 5.01). P<0.05 was considered significant.
RESULTS
Slow Progressive Conduction Defects and Heart Enlargement in Nkx2-5 KNOCKOUT MICE from 2 Weeks of Age
Recently, we generated a mouse model to delete the Nkx2-5 gene by tamoxifen injection15 by mating mice homozygous for floxed-Nkx2-5 alleles8 and heterozygous for the Cre-ER transgene under the control of the CMV enhancer and the chicken β-globin promoter.21 Tamoxifen injection within 24 h before birth induced rapid progressive conduction and contraction defects within 4 days, which leads to premature death.15
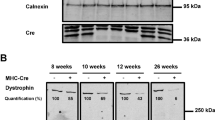
In this study, tamoxifen was injected at 2 weeks of age for 4 consecutive days. Time course studies at multiple time points are shown in Figure 1a. Measurements of heart weight/tibial length and telemetry ECG recordings of Nkx2-5 knockout mice were determined at multiple time points from 3.5 to 24 weeks of age (1.5 and 22 weeks from the initiation of tamoxifen injections, respectively). Deletion of the Nkx2-5 gene, followed by a reduction of Nkx2-5 mRNA (Figure 1b) and proteins (Figure 1c), was demonstrated 1 day after four consecutive tamoxifen injections (approximately 2.5 weeks of age), as well as 3 days (approximately 3 weeks of age) and 6 days (approximately 3.5 weeks of age) after injections. The reduction of Nkx2-5 mRNA continued up to 24 weeks of age by tamoxifen injections at 2 weeks of age, as demonstrated in northern blotting (Figure 1d).

Time course study of tamoxifen-inducible targeting of Nkx2-5 beginning at 2 weeks of age. (a) Experimental time course of Nkx2-5 knockout beginning at 2 weeks of age. (b) Northern blotting demonstrated a nearly complete loss of Nkx2-5 mRNA from 2.5 weeks (immediate after the completion of four consecutive tamoxifen injections) to 3.5 weeks in flox/flox/Cre compared with flox/flox mice. (c) Western blotting demonstrated a marked reduction of Nkx2-5 proteins (arrowheads) from 2.5 to 3.5 weeks of age in flox/flox/Cre mice. A nonspecific band was detected at 2.5 weeks of age (marked with *). (d) Northern blotting demonstrated a reduced expression of Nkx2-5 in flox/flox/Cre compared with flox/flox mice at 3.5 and 24 weeks of age after tamoxifen injections. fl, flox; wks, weeks.
Telemetry ECG recordings of Nkx2-5 knockout mice and tamoxifen-injected control mice (flox/flox or flox/wild/Cre) were performed at multiple time points from 3.5 to 24 weeks of age (1.5 and 22 weeks after tamoxifen injections, respectively) (Table 1). Nkx2-5 knockout mice first showed PR prolongation at 9 weeks of age, accompanied by an intermittent 2° AV block. The PR interval was progressively prolonged, and wide QRS was observed at 24 weeks of age (Table 1). With the exception of one mouse at 24 weeks of age with an intermittent 2° AV block, heterozygous Nkx2-5 knockout mice (flox/wild/Cre) did not show PR prolongation or wide QRS (Table 1). Examples of telemetry ECG recordings normal at 3.5 weeks of age and with a demonstration of the 2° AV block and wide QRS at 24 weeks of age in Nkx2-5 knockout hearts are shown in Figure 2a.

AV block and heart enlargement in Nkx2-5 knockout mice. (a) Examples of telemetry ECG recording at 3.5 and 24 weeks of age. At 24 weeks of age, telemetry ECG recording from flox/flox mouse showed normal sinus rhythm (left panel); however, that from flox/flox/Cre showed a 2° AV block (right panel). See Table 1. (b) Heart weight/tibial length in Nkx2-5 knockout mice compared with that in control mice (flox/flox or flox/wild/Cre), with a significant increase in heart weight/tibial length in Nkx2-5 knockout mice at 12 and 24 weeks of age. (c and d) Representative imaging using an M-mode ultrasound microscope and echocardiographic indices of control and Nkx2-5 knockout mice of approximately 24 weeks of age (mean±s.e.) are shown. SCL, sinus cycle length (beta-to beat heart rate); BW, body weight; HR, heart rate; LVDd, left ventricular diastolic dimension; LVDs, left ventricular systolic dimension; %FS, percentage of left ventricular fractional shortening.
Heart weight/tibial length, an indicator of heart enlargement, was slightly but significantly increased in mice with Nkx2-5 knockout beginning at 12 weeks of age compared with that in tamoxifen-injected control mice (flox/flox or flox/wild/Cre) (Figure 2b). A reduction of cardiac contractile performance (% fraction shortening) and an increase of left ventricular systolic dimension (LVDs) were observed in Nkx2-5 knockout mice at 24 weeks of age, using 40 MHz ultrasound imaging (Figures 2c and d).
Taken together, mice with loss of Nkx2-5 from 2 weeks of age show conduction and contraction defects. Although this phenotype is qualitatively similar to the phenotype observed with the perinatal loss of Nkx2-5,15 disease progression and severity are markedly distinct despite using a genetically identical mouse model. For instance, when tamoxifen was administered at 2 weeks of age, no phenotype was apparent over a time span of 1.5 weeks after tamoxifen injection (3.5 weeks of age), in contrast to perinatal tamoxifen administration.15 In addition, mice with loss of Nkx2-5 from 2 weeks of age, survive over 1.5 years of age, despite the presence of PR prolongation, wide QRS and a slight increase in heart weight/tibial length (data not shown), whereas mice with perinatal loss of Nkx2-5 die prematurely.15
Reduced Expression of Nkx2-5 Protein in Cardiomyocytes Accompanied by Small AV Node
Immunostaining confirmed a decreased Nkx2-5 expression in Nkx2-5 knockout ventricles at 3.5 weeks of age (Figure 3a, left panels). Despite a slight heart enlargement in Nkx2-5 knockout mice at 24 weeks of age, interstitial fibrosis was not apparent in ventricles (Figure 3a, right panels). In adjacent tissue sections, including the acetylcholine esterase-positive AV node, positive Nkx2-5 staining was shown in the control heart, but not in the Nkx2-5 knockout heart (Figure 3b). Although formation of the AV node is completed before birth, whole-mount acetylcholine esterase staining showed that the AV nodal surface area size was smaller at 24 weeks of age in hearts with a loss of Nkx2-5 beginning at 2 weeks of age (Figures 3c and d). This observation is similar to previous studies using ventricular-specific deletion of Nkx2-5 from the embryonic stage in MLC2v-Cre mice and in perinatal Nkx2-5 knockout mice;8 however, AV nodal fibrosis was not apparent in mice with Nkx2-5 knockout in perinatal15 and at 2 weeks of age (Figure 3e).

Nkx2-5 protein expression in contractile myocardium and AV node. (a) Coimmunostaining of Nkx2-5 and sarcomeric actinin demonstrates a reduced expression of Nkx2-5 protein in ventricles dissected from Nkx2-5 mice at 3.5 weeks of age (left panels). Heart sections from 24-week-old mice stained with Masson's trichrome show no apparent interstitial fibrosis in Nkx2-5 knockout mice compared with that in control mice (right panels). Bars=100 μm. (b) Tissue sections of 3.5-week-old mouse heart show that AV node is positive for acetylcholine esterase staining (AchE) both in flox/flox and flox/flox/Cre mice. The adjacent tissue sections including AV node stained positive with an anti-Nkx2-5 antibody (green) in flox/flox mice but was negative in flox/flox/Cre mice. Sarcomeric actinin staining is shown in red. Bars=100 μm. (c) Whole-mount acetylcholine esterase staining demonstrates AV node (brown staining, arrowheads) both in flox/flox and flox/flox/Cre mice at 3.5 and 24 weeks of age. Bars=1 mm. (d) Surface area positive for acetylcholine esterase compared between flox/flox and flox/flox/Cre mice at 3.5 and 24 weeks of age (mean±s.e.). *P<0.05. (e) No apparent fibrosis was observed in Nkx2-5 knockout AV node analyzed by Masson's trichrome staining at 24 weeks of age. The AV nodal area is marked with arrows. Bars in low magnifications=200 μm. Bars in high magnifications=50 μm. (f) Nuclear density (number of nuclei/mm2) in flox/flox and flox/flox/Cre mice at 24 weeks of age. The total nuclear number counted was from a total of three different flox/flox or flox/flox/Cre mice. (g) HE-stained longitudinal sections of cardiomyocytes in AV node (left upper panels) and LV upper free wall (left bottom panels) were utilized for measurement of cell width at the nuclei level at 3.5 and 24 weeks of age. Bars=20 μm. (h) AV nodal cell width (left panel) and LV cell width (right panel) compared between flox/flox and flox/flox/Cre mice at 3.5 and 24 weeks of age (mean±s.e.). *P<0.05. The total cell number counted was from a total of three different flox/flox or flox/flox/Cre mice at 3.5 weeks and four different flox/flox or flox/flox/Cre mice at 24 weeks. MV, mitral valve; Ao, aorta; IVS, interventricular septum.
In addition, nuclei in the AV node appeared more condensed and the nuclear number/mm2 was significantly increased in the AV node in Nkx2-5 knockout vs control hearts at 24 weeks of age (Figure 3e and f). To examine whether this could be because of reduced cardiomyocyte cell size in the AV node of Nkx2-5 knockout mice, cell diameter was measured in the longitudinal section at the position of nuclei. The AV nodal cell width in control flox/flox mice was increased from 3.5 to 24 weeks of age, but was not increased in flox/flox/Cre mice, leading to a smaller AV nodal cell width in Nkx2-5 knockout vs control mice at 24 weeks of age (Figure 3g, upper panels and h, left panel).
In contrast, cell width in non-AV nodal myocytes in the left ventricle was increased between 3.5 and 24 weeks of age in both Nkx2-5 knockout and control mice, and was slightly, but not significantly, wider in Nkx2-5 knockout at 24 weeks of age (Figure 3g, lower panels and h, right panel). This observation was further examined using isolated cardiomyocyte measurements that showed a significant increase in the mean surface area, long axis and short axis length in Nkx2-5 knockout myocytes (Figures 4a–c). Taken together, Nkx2-5 knockout hearts at 24 weeks of age showed ventricular myocyte hypertrophy, and a reduction in cardiomyocyte cell width in the AV node.

Increased cell size and Ca2+-handling defects in cardiomyocytes isolated from Nkx2-5 knockout heart. (a) Representative images of cardiomyocytes isolated from flox/flox (left) and flox/flox/Cre mice (right) at 24 weeks of age. An increased cell size was shown in flox/flox/Cre mice. Bars=100 μm. (b) Cell area (μm2), short axis (μm) and long axis (μm) of cardiomyocytes isolated from flox/flox or flox/flox/Cre mice (n=300 from 3 mice). (c) Distribution of the cell area of cardiomyocytes isolated from flox/flox and flox/flox/Cre mice. (d) Measurements of cardiac contraction and simultaneous Ca2+ transients in isolated cardiomyocytes from control flox/flox (white bars) or Nkx2-5 knockout (flox/flox/Cre) mice (black bars). Summarized data (mean±s.e.) demonstrate that Nkx2-5 knockout cardiomyocytes show decreased Ca2+ amplitude and systolic fluorescence ratio, but preserved %fractional shortening, +dL/dT, −dL/dT and diastolic fluorescence ratio. *P<0.05. (e) Increase of Ca2+ concentration in superfusate from 1.2 to 2.5 mM increased %fractional shortening both in control and Nkx2-5 knockout cardiomyocytes, but was less in Nkx2-5 knockout cardiomyocytes compared with that in control cardiomyocytes.
Nkx2-5 Knockout Ventricular Cardiomyocytes are Morphologically and Functionally Compensated
At 24 weeks of age, cardiomyocytes isolated from mice with Nkx2-5 knockout from 2 weeks of age did not show significant differences in fractional shortening, +dL/dT (rate of contraction) and −dL/dT (rate of relaxation) compared with cells from control mice at a Ca2+ superfusate concentration of 1.2 mM. (Figure 4d). Notably, the Ca2+ transient amplitude and systolic fluorescence ratio were significantly lower in Nkx2-5 knockout cardiomyocytes (Figure 4d), which is similar to perinatal Nkx2-5 knockout cardiomyocytes with a reduction of ryanodine receptor 2.15 When the Ca2+ superfusate concentration was increased from 1.2 to 2.5 mM, the increase in fractional shortening was blunted in Nkx2-5 knockout cardiomyocytes compared with that in control cardiomyocytes (Figure 4e). These results indicate that cardiomyocytes from Nkx2-5 knockout hearts were morphologically and functionally compensated despite reduced Ca2+handling, and a reduction in contraction appeared only under conditions of increased demand.
Slow Reduction of Nkx2-5 Downstream Targets in Hearts with Loss of Nkx2-5 from 2 Weeks of Age
The expressions of cardiac hypertrophic markers, ANF and BNP are regulated by Nkx2-5 shown in embryos7, as well as at PD4 in the perinatal loss of Nkx2-5 (Figure 5a). In hearts from mice with Nkx2-5 knockout from 2 weeks of age, the expression of ANF and BNP was also downregulated early after tamoxifen injections at 3.5 weeks of age (1.5 weeks after tamoxifen injection) and further decreased at 6 weeks of age (4 weeks after tamoxifen injection) compared with that in control flox/flox hearts (Figure 5b).

The expression of Nkx2-5 target genes in Nkx2-5 knockout hearts. (a) Northern blotting shows reduction of ANF and BNP mRNA and induction of βMHC mRNA in perinatal Nkx2-5 knockout hearts at PD4. (b) Northern blotting shows reduction of ANF and BNP mRNA at 3.5 and 6 weeks of age and induction of βMHC mRNA at 6 weeks of age in Nkx2-5 knockout hearts when tamoxifen was injected from 2 weeks of age. (c) Real-time RT-PCR shows fold difference (Nkx2-5 knockout vs control) of mRNA of cardiac MLCK, Nav1.5(α) and RyR2 at 3.5 weeks, 9 weeks and 6 months of age. MLCK, myosin light chain kinase; RyR2, ryanodine receptor 2; KO, knockout.
In contrast, the expression of another hypertrophic marker, β-MHC, was induced 4 days after tamoxifen injection in hearts with a perinatal loss of Nkx2-5 (Figure 5a), whereas it was barely detected at 3.5 weeks of age in mice with Nkx2-5 knockout from 2 weeks of age, but was upregulated at 6 weeks of age (Figure 5b). Thus, the delayed downregulation of ANF and BNP, as well as the delayed upregulation of βMHC, in hearts from mice with Nkx2-5 knockout from 2 weeks of age contrasts with the rapid changes in expression within 4 days after tamoxifen injection in hearts with a perinatal loss of Nkx2-5. At 24 weeks of age, when marked cardiomyocyte hypertrophy was demonstrated (Figures 4a–c), the expression of ANF was increased to the level of control (fold difference flox/flox/Cre vs flox/flox, means±s.e., 1.11±0.02, n=2); however, the expression of BNP remained reduced (0.27±0.00, n=2) and that of βMHC remained upregulated (5.12±0.39, n=2).
In hearts with perinatal loss of Nkx2-5, a marked reduction (80–90%) of three additional gene products important for cardiac conduction and contraction, including the Na+ channel, Nav1.5(α), ryanodine receptor 2 and cardiac MLCK, was also demonstrated by PD12.15, 16 A reduction in expression was also observed in hearts from mice with loss of Nkx2-5 beginning at 2 weeks of age (Figure 5c); however, the magnitude of the reduction was less and the time course was prolonged in hearts with Nkx2-5 knockout from 2 weeks of age compared with those with a perinatal loss of Nkx2-5. Taken together, a slower progression and decreased magnitude of reduction of Nkx2-5 downstream targets characterize the phenotype of hearts with loss of Nkx2-5 from 2 weeks of age. This observation is one potential mechanism explaining the slower disease progression.
DISCUSSION
Nkx2-5 is a critical factor in fetal cardiac development when cardiomyocytes are proliferating as demonstrated in various studies using mouse embryos or stem cells.13, 14 Nkx2-5 is also important in perinatal hearts, as demonstrated by rapid conduction and contraction defects observed within 4 days after the deletion of Nkx2-5 following tamoxifen injection, leading to premature death.15 The loss of Nkx2-5 from 2 weeks of age results in an overall similar phenotype, but with a distinct disease progression and severity despite using a genetically identical mouse model with a 2-week difference in the administration of tamoxifen injection. For example, the perinatal loss of Nkx2-5 results in premature death, but the loss of Nkx2-5 from 2 weeks of age does not. An apparent phenotype emerges within 4 days with the perinatal loss of Nkx2-5 vs over 7 weeks with the loss beginning at 2 weeks of age.
With respect to the pathogenesis of human congenital AV block and occasional LV dysfunction associated with NKX2-5 mutations, our previous15 and current studies reveal that Nkx2-5 is necessary for proper conduction and contraction postnatally, but is more critical in the perinatal heart and is necessary for survival in mice. Haploinsufficiency (loss of one allele) of Nkx2-5 is considered to be an underlying cause of human congenital heart disease as shown in several germline heterozygous-null Nkx2-5 mice.26, 27, 28, 29 Although substantially different degrees of defects among studies were reported, PR prolongation accompanied with wide QRS was detected as early as 7 weeks of age.28 When the Nkx2-5 gene was deleted after 2 weeks of age, heterozygous-null mice did not apparently develop heart enlargement, PR prolongation or wide QRS by 24 weeks of age, with the exception of one mouse with an intermittent 2° AV block at 24 weeks of age.
We initiated this study on the basis of the hypothesis that cardiomyocytes before and after G1 cell-cycle arrest may demonstrate morphological, metabolic and gene expression changes with a re-organization of nuclear architecture and chromatin structure.19, 20 In fact, we found a global increase of DNA methylation in hearts within 2 weeks after birth using 5-methylcytosine staining (data not shown).30 The question remains as to how an increase in broad DNA methylation relates to slower and milder phenotypes in mice with loss of Nkx2-5 from 2 weeks age. It might be argued that a compact chromatin DNA structure due to DNA methylation may delay transcriptional re-regulation after the loss of Nkx2-5 from 2 weeks of age. Another mechanism is that other transcription factors might compensate for the absence of Nkx2-5 after 2 weeks of age. The mRNA half-life of Nkx2-5 targets after 2 weeks of age might be substantially longer compared with that at the neonatal stage.
To our knowledge, a limited number of cardiac transcription factors have been studied using postnatal-specific gene deletion. The loss of serum responsive factor (SRF) from 2 months of age leads to lethal heart failure 10 weeks after tamoxifen injection.31 Although a 70% reduction of the SRF downstream target, α-actin, was demonstrated 5 days after tamoxifen injections, its expression did not change appreciably 30 and 60 days after tamoxifen injection. The expression of other SRF targets, such as vinculin and zyxin, demonstrated in ES cells, as well as in Nkx2-5, MEF2C, GATA4 and TEF1, demonstrated after the loss of SRF in embryonic hearts (floxed-SRF mice crossed with βMHC-Cre transgenic mice), was unchanged or fluctuated 5, 30 and 60 days after tamoxifen injection.31 Thus, this study also demonstrated a differential regulation of transcription in adult vs proliferating cardiomyocytes. Additional studies using postnatal-specific gene targeting of cardiac transcription factors will reveal whether the developmental stage-dependent effects are a general property of cardiac transcription factors.
Nkx2-5 has been shown to be involved in cardiac hypertrophy;32, 33, 34 however, this study shows that without Nkx2-5, ventricular cardiomyocytes have compensatory hypertrophy at 24 weeks of age. Thus, Nkx2-5 may not be critically important for adult ventricular cardiomyocyte hypertrophy as was suggested in a recent review.35 In contrast, Nkx2-5 might be important for cellular enlargement in AV nodal cardiomyocytes during postnatal development, as demonstrated in this study, which are not involved in contraction nor are they affected by compensatory hypertrophic stimuli. It is noteworthy that defects in AV bundle development during embryonic stages have been shown in heterozygous Nkx2-5 knockout mice.29 Morphological changes in ventricular conduction systems, including AV bundle, remain to be analyzed in postnatal Nkx2-5 knockout hearts.
Cardiomyocyte hypertrophy is usually accompanied by an increase in the expression of ANF and BNP. The expression of those markers in hypertrophic cardiomyocytes with a loss of their upstream regulatory transcription factor, Nkx2-5, is somewhat difficult to interpret and is interesting. We found that soon after the loss of Nkx2-5 (3.5 and 6 weeks of age, Figure 5b), the expression of ANF and BNP was decreased because of the loss of their transactivator, Nkx2-5. At 24 weeks of age, when cardiomyocyte hypertrophy was evident, ANF expression was increased to the normal level, but BNP expression remained downregulated. One potential interpretation of the time-dependent changes in ANF expression (downregulation soon after the loss of Nkx2-5 and normalization at 24 weeks of age), which are highly induced and overcome the loss of Nkx2-5 function in the transcription of the ANF gene, would be because of an Nkx2-5-independent regulatory mechanism at 24 weeks of age, resulting in an increase in ANF expression to the normal control level.
In summary, we demonstrated that Nkx2-5 actively regulates a critical set of genes in postnatal cardiomyocytes to maintain proper cardiac function. However an age-related phenotypic difference after the loss of Nkx2-5 between 2 weeks of age vs perinatal stage was identified, which is accompanied by a slower and smaller reduction of several critical Nkx2-5 responsive genes.
References
Harvey RP . NK-2 homeobox genes and heart development. Dev Biol 1996;178:203–216.
Harvey RP, Rosenthal N . Heart Development. Academic Press: San Diego, CA, USA, 1999.
Lints TJ, Parsons LM, Hartley L, et al. Nkx-2.5: a novel murine homeobox gene expressed in early heart progenitor cells and their myogenic descendants. Development 1993;119:419–431.
Komuro I, Izumo S . Csx: a murine homeobox-containing gene specifically expressed in the developing heart. Proc Natl Acad Sci USA 1993;90:8145–8149.
Kasahara H, Bartunkova S, Schinke M, et al. Cardiac and extracardiac expression of Csx/Nkx2.5 homeodomain protein. Circ Res 1998;82:936–946.
Lyons I, Parsons LM, Hartley L, et al. Myogenic and morphogenetic defects in the heart tubes of murine embryos lacking the homeo box gene Nkx2-5. Genes Dev 1995;9:1654–1666.
Tanaka M, Chen Z, Bartunkova S, et al. The cardiac homeobox gene Csx/Nkx2.5 lies genetically upstream of multiple genes essential for heart development. Development 1999;126:1269–1280.
Pashmforoush M, Lu JT, Chen H, et al. Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell 2004;117:373–386.
Schott JJ, Benson DW, Basson CT, et al. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998;281:108–111.
Benson DW, Silberbach GM, Kavanaugh-McHugh A, et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest 1999;104:1567–1573.
Kasahara H, Benson DW . Biochemical analyses of eight NKX2.5 homeodomain missense mutations causing atrioventricular block and cardiac anomalies. Cardiovasc Res 2004;64:40–51.
Konig K, Will JC, Berger F, et al. Familial congenital heart disease, progressive atrioventricular block and the cardiac homeobox transcription factor gene NKX2.5: identification of a novel mutation. Clin Res Cardiol 2006;95:499–503.
Sachinidis A, Fleischmann BK, Kolossov E, et al. Cardiac specific differentiation of mouse embryonic stem cells. Cardiovasc Res 2003;58:278–291.
Puceat M . Rb and LEK1: a ‘pas de deux’ in cardiogenesis. Cell Cycle 2005;4:1030–1032.
Briggs LE, Takeda M, Cuadra AE, et al. Perinatal loss of Nkx2-5 results in rapid conduction and contraction defects. Circ Res 2008;103:580–590.
Chan JY, Takeda M, Briggs LE, et al. Identification of cardiac-specific myosin light chain kinase. Circ Res 2008;102:571–580.
Anversa P, Leri A, Kajstura J . Cardiac regeneration. J Am Coll Cardiol 2006;47:1769–1776.
Ahuja P, Sdek P, MacLellan WR . Cardiac myocyte cell cycle control in development, disease, and regeneration. Physiol Rev 2007;87:521–544.
Oberdoerffer P, Sinclair DA . The role of nuclear architecture in genomic instability and ageing. Nat Rev Mol Cell Biol 2007;8:692–702.
Rowat AC, Lammerding J, Herrmann H, et al. Towards an integrated understanding of the structure and mechanics of the cell nucleus. Bioessays 2008;30:226–236.
Hayashi S, McMahon AP . Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 2002;244:305–318.
Wakimoto H, Kasahara H, Maguire CT, et al. Developmentally modulated cardiac conduction failure in transgenic mice with fetal or postnatal overexpression of DNA nonbinding mutant Nkx2.5. J Cardiovasc Electrophysiol 2002;13:682–688.
Scherrer-Crosbie M, Ullrich R, Bloch KD, et al. Endothelial nitric oxide synthase limits left ventricular remodeling after myocardial infarction in mice. Circulation 2001;104:1286–1291.
Janssens S, Pokreisz P, Schoonjans L, et al. Cardiomyocyte-specific overexpression of nitric oxide synthase 3 improves left ventricular performance and reduces compensatory hypertrophy after myocardial infarction. Circ Res 2004;94:1256–1262.
Minhas KM, Saraiva RM, Schuleri KH, et al. Xanthine oxidoreductase inhibition causes reverse remodeling in rats with dilated cardiomyopathy. Circ Res 2006;98:271–279.
Biben C, Weber R, Kesteven S, et al. Cardiac septal and valvular dysmorphogenesis in mice heterozygous for mutations in the homeobox gene Nkx2-5. Circ Res 2000;87:888–895.
Tanaka M, Berul CI, Ishii M, et al. A mouse model of congenital heart disease: cardiac arrhythmias and atrial septal defect caused by haploinsufficiency of the cardiac transcription factor Csx/Nkx2.5. Cold Spring Harb Symp Quant Biol 2002;67:317–325.
Jay PY, Harris BS, Maguire CT, et al. Nkx2-5 mutation causes anatomic hypoplasia of the cardiac conduction system. J Clin Invest 2004;113:1130–1137.
Moskowitz IP, Kim JB, Moore ML, et al. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell 2007;129:1365–1376.
Sedmera D, Pexieder T, Vuillemin M, et al. Developmental patterning of the myocardium. Anat Rec 2000;258:319–337.
Parlakian A, Charvet C, Escoubet B, et al. Temporally controlled onset of dilated cardiomyopathy through disruption of the SRF gene in adult heart. Circulation 2005;112:2930–2939.
Thompson JT, Rackley MS, O'Brien TX . Upregulation of the cardiac homeobox gene Nkx2-5 (CSX) in feline right ventricular pressure overload. Am J Physiol 1998;274 (5 Part 2):H1569–H1573.
Saadane N, Alpert L, Chalifour LE . Expression of immediate early genes, GATA-4, and Nkx-2.5 in adrenergic-induced cardiac hypertrophy and during regression in adult mice. Br J Pharmacol 1999;127:1165–1176.
Bar H, Kreuzer J, Cojoc A, et al. Upregulation of embryonic transcription factors in right ventricular hypertrophy. Basic Res Cardiol 2003;98:285–294.
Oka T, Xu J, Molkentin JD . Re-employment of developmental transcription factors in adult heart disease. Semin Cell Dev Biol 2007;18:117–131.
Acknowledgements
We greatly appreciate E Chan and P Sayeski for their valuable suggestions and technical support. This study was supported by the National Institutes of Health (HL081577 to HK) and the American Heart Association-National SDG grant (0035258N to HK).
Author information
Authors and Affiliations
Corresponding author
Additional information
Duality of interest
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Takeda, M., Briggs, L., Wakimoto, H. et al. Slow progressive conduction and contraction defects in loss of Nkx2-5 mice after cardiomyocyte terminal differentiation. Lab Invest 89, 983–993 (2009). https://doi.org/10.1038/labinvest.2009.59
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.2009.59
Keywords
This article is cited by
-
Cardiac MLC2 kinase is localized to the Z-disc and interacts with α-actinin2
Scientific Reports (2019)
-
Transcriptional activator DOT1L putatively regulates human embryonic stem cell differentiation into the cardiac lineage
Stem Cell Research & Therapy (2018)
-
Transcriptional regulation of the cardiac conduction system
Nature Reviews Cardiology (2018)
-
Maternal NO2 exposure induces cardiac hypertrophy in male offspring via ROS-HIF-1α transcriptional regulation and aberrant DNA methylation modification of Csx/Nkx2.5
Archives of Toxicology (2018)
-
Mutation of a common amino acid in NKX2.5 results in dilated cardiomyopathy in two large families
BMC Medical Genetics (2016)
