
Abstract
The etiology of idiopathic short stature (ISS) and Leri–Weill dyschondrosteosis (LWD) in European patients is known to include SHOX mutations and copy-number variations (CNVs) involving SHOX and/or the highly evolutionarily conserved non-coding DNA elements (CNEs) flanking the gene. However, the frequency and types of SHOX abnormalities in non-European patients and the clinical importance of mutations in the CNEs remains to be clarified. Here, we performed systematic molecular analyses of SHOX for 328 Japanese patients with ISS or LWD. SHOX abnormalities accounted for 3.8% of ISS and 50% of LWD cases. CNVs around SHOX were identified in 16 cases, although the ~47 kb deletion frequently reported in European patients was absent in our cases. Probably damaging mutations and benign/silent substitutions were detected in four cases, respectively. Although CNE-linked substitutions were detected in 15 cases, most of them affected poorly conserved nucleotides and were shared by unaffected individuals. These results suggest that the frequency and mutation spectrum of SHOX abnormalities are comparable between Asian and European patients, with the exception of a European-specific downstream deletion. Furthermore, this study highlights the clinical importance and genetic heterogeneity of the SHOX-flanking CNVs, and indicates a limited clinical significance of point mutations in the CNEs.
Similar content being viewed by others

Introduction
SHOX (NM_000451.3) is located in the short arm pseudoautosomal region of the sex chromosomes (PAR1), and encodes a homeobox-containing transcription factor that plays a critical role in skeletal growth.1 SHOX haploinsufficiency leads to idiopathic short stature (ISS; OMIM #300582) without skeletal malformations and Leri–Weill dyschondrosteosis (LWD; OMIM #127300) characterized by Madelung deformity.1, 2, 3 Madelung deformity is a cluster of anatomical changes in the wrist including bowing of the radius and dorsal dislocation of the distal ulna, which can result in wrist pain, deformation and/or limited joint mobility.2, 3 Genetic defects underlying SHOX haploinsufficiency include several mutations in the coding region and various copy-number variations (CNVs) in PAR1.2, 4, 5, 6, 7, 8, 9, 10, 11 The ISS/LWD-associated CNVs are predicted to involve SHOX exons and/or cis-acting enhancers. Although the precise positions of the SHOX enhancers remain to be determined, they likely reside within the highly evolutionarily conserved non-coding DNA elements (CNEs) around the gene.10, 11, 12, 13, 14 Previous studies have identified seven CNEs (CNE-5, CNE-3, CNE-2, CNE4, CNE5, evolutionarily conserved region (ECR) 1 and evolutionarily conserved sequence (ECS) 4/CNE9) that exert in vitro or in vivo cis-regulatory activity.10, 11, 12, 13, 14 In vitro assays confirmed physical interaction between these CNEs and the SHOX promoter.10, 15
So far, mutation screening and copy-number analyses of SHOX have been performed mostly for patients of European origin.4, 5, 6, 7, 8, 9, 10, 16, 17, 18, 19, 20 Previous studies indicated that SHOX haploinsufficiency is primarily caused by submicroscopic CNVs in PAR1, and accounts for 2–17% of ISS cases and 35–100% of LWD cases.4, 5, 6, 7, 8, 9, 10, 16, 17, 18, 19, 20 However, the applicability of these findings to non-European populations remains unclear. For example, although a ~47 kb deletion in the SHOX downstream region was frequently identified in European patients,10, 19, 20 it is unknown whether this CNV is shared by patients of other ethnicities. More importantly, there is no report of sequence analysis of the CNEs in the patients with ISS or LWD. Thus, the clinical importance of point mutations in the known CNEs has yet to be studied. To address these unsolved issues, we performed systematic molecular analysis of SHOX in a large cohort of Japanese patients.
Materials and methods
Subjects
The study was approved by the Institutional Review Board Committee at the National Center for Child and Development, and performed after obtaining informed consent from the participants or their parents.
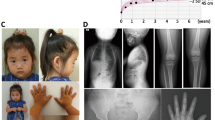
The study group consisted of 328 Japanese patients with short stature (164 males and 164 females; aged 0.5–17.9 years). The patients satisfied the following conditions: (i) referred to our pediatric endocrinology clinics between March 2013 and November 2015 for evaluation of short stature; (ii) short stature with standard deviation scores of <−2.0; (iii) no chronic diseases, such as growth hormone deficiency, congenital heart disease, achondroplasia or thyroid disease that may affect growth; and (iv) lack of cytogenetically detectable chromosomal abnormalities. The patients underwent radiological examinations of the hand. We examined the presence or absence of Madelung deformity-compatible features including narrowing of the ulnar portion of the distal radial physis, anterior bowing of the radial shaft and dorsal subluxation of the ulnar head.3 Of the 328 patients, 16 with radiologically recognizable Madelung deformity were diagnosed with LWD, whereas the remaining 312 were diagnosed with ISS. Four patients were included from our previous study.21 As controls, we used genomic DNA samples obtained from 100 healthy Japanese adults with normal height (50 males and 50 females).
Copy-number analysis
All patients were subjected to copy-number analysis. Genomic DNA was extracted from peripheral leukocytes. CNVs in PAR1 were analyzed by multiplex ligation-dependent probe amplification using a commercially available kit (SALSA P018-G1, MRC-Holland, Amsterdam, Netherlands) and further characterized by array-based comparative genomic hybridization using a custom-made microarray (8 × 60 k format, Agilent Technologies, Santa Clara, CA, USA). To exclude non-pathogenic variations, we referred to the Database of Genomic Variants (http://projects.tcag.ca/variation/).
Sequence analysis
Of the 312 patients with no pathogenic CNVs, 309 (300 ISS and 9 LWD) were subjected to sequence analysis of SHOX exons; 3 patients were excluded from this analysis because of insufficient amounts of DNA samples. We also carried out sequence analysis of the 7 CNEs for 83 patients (76 ISS and 7 LWD) whose DNA samples were sufficient for this experiment.
DNA fragments corresponding to the target regions were amplified by multiplex-PCR or by using the Haloplex system (Agilent Technologies, Santa Clara, CA, USA) and sequenced on a MiSeq next-generation sequencer (Illumina, San Diego, CA, USA). The highly polymorphic oligonucleotide repeats in the CNEs were excluded from sequence analysis. Nucleotide changes indicated by next-generation sequencing were confirmed by the Sanger method. Primer sequences are available on request. Nucleotide substitutions whose allelic frequency in the databases (dbSNP, http://www.ncbi.nlm.nih.gov/snp/; or 1000 Genome Browser, http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) was more than 0.05 were excluded as common polymorphisms.
The functional outcomes of missense substitutions were predicted by in silico analysis using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/). To examine the population frequency of exonic substitutions, we referred to the Exome Aggregation Consortium Browser (http://exac.broadinstitute.org/) and the Human Genetic Variation Browser (http://www.genome.med.kyoto-u.ac.jp/SnpDB). To evaluate the population frequency of novel substitutions, we analyzed DNA samples of control individuals.
To assess the functional significance of nucleotide changes in the CNEs, we examined whether these substitutions were located within the putative enhancer sequences. We referred to ENCODE Broad Chromatin State Segmentation in the UCSC browser (http://genome.ucsc.edu/). The evolutionary conservation of the affected nucleotides was analyzed using the UCSC browser.
Results
Copy-number analysis
Submicroscopic CNVs in PAR1 were identified in 16 cases (cases 1–16; Figure 1 and Supplementary Figures 1 and 2). These CNVs consisted of 11 deletions and 5 duplications. The deletion in case 1 extended beyond PAR1, whereas CNVs in the remaining cases were located within PAR1. The deletion in case 9 affected a genomic interval downstream of the known CNEs. The ~47 kb deletion common in European patients,10, 19, 20 was absent in our cases. The breakpoints of the CNVs in cases 2, 3, 5, 12 and 14 were located within or close to repetitive sequences (Supplementary Figure 2).

Copy-number variations (CNVs) identified in cases 1–16. The upper panel shows the names of multiplex ligation-dependent probe amplification (MLPA) probes. Probes of the SHOX exons are shaded in purple, and those of the putative enhancer regions, that is, highly evolutionarily conserved non-coding DNA elements (CNEs), evolutionally conserved region (ECR), or evolutionally conserved sequence (ECS), are shaded in blue or yellow. Probes in the pseudoautosomal region 1 are indicated with an arrow. The green and red lines depict copy-number loss and gain, respectively. The dotted lines indicate the copy-number unknown regions. p-ter: short arm telomere; cen, centromere.
Sequence analysis
Sequence analysis of SHOX identified eight intragenic substitutions in eight cases (cases 17–24; Table 1 and Figure 2). Of these, five were missense substitutions, and three were silent (synonymous) changes. These eight substitutions were absent or extremely rare in the general population. Four of the five missense substitutions were assessed as ‘probably damaging’, whereas p.Ser46Phe in case 21 was scored as ‘benign’. The substitutions in cases 17–19 were located within the homeobox.

Nucleotide substitutions identified in cases 17–39. (a) Genomic structure of SHOX and its flanking region. Positions refer to the Human Genome (hg 19; NCBI Build 37). The white and purple boxes indicate the untranslated and translated exons, respectively. The striped boxes depict the homeobox. The yellow and light blue boxes denote the putative upstream and downstream enhancer regions, respectively. Probably damaging mutations and benign/silent substitutions are indicated by dark blue and green arrows, respectively. Substitutions in the putative enhancer regions are depicted by light blue arrows. (b) Electrochromatographs of the substitutions. Mutated nucleotides are indicated by arrows. Substitutions indicated by a, b, and c were identified in multiple patients. p-ter: short arm telomere; cen, centromere.
Sequence analysis of the known CNEs detected 11 substitutions in 15 cases (cases 25–39; Table 1 and Figure 2). Of these, five were rare polymorphisms, and the others were first identified herein. Most of the novel substitutions were shared by the control individuals. Substitutions in CNE-5 and CNE5 resided within putative enhancer sequences (Supplementary Figure 3). Most of the altered nucleotides were not highly conserved among species (Supplementary Figure 4).
Clinical features of patients with SHOX abnormalities
Cases 1–16 with CNVs comprised ten ISS and six LWD patients, whereas cases 17–20 with probably damaging mutations included two ISS and two LWD patients (Table 2). Thus, apparent SHOX abnormalities accounted for 12 of 312 (3.8%) ISS patients and eight of 16 (50%) LWD patients. Cases with benign or silent substitutions (cases 21–24) and CNE-linked substitutions (cases 25–39) were all ISS.
Discussion
Systematic molecular analysis identified probably pathogenic SHOX abnormalities in 3.8 and 50% of Japanese ISS and LWD cases, respectively. These results indicate that the frequency of SHOX haploinsufficiency is comparable between Japanese and European patients. On the other hand, none of our patients carried the well-known ~47 kb downstream deletion,10, 19, 20 indicating that this CNV is a European-specific founder mutation. The lack of SHOX abnormalities in seven LWD patients provides further evidence for the allelic heterogeneity of LWD. As mutations in NPR2, a causative gene of Maroteaux-type acromesomelic dysplasia, have recently been identified in patients with LWD-compatible clinical features,22 such mutations may be present in some of our patients with normal SHOX.
Submicroscopic CNVs in PAR1 were identified in cases 1–16. Our results highlight the importance of PAR1-linked CNVs as a cause of SHOX haploinsufficiency. Furthermore, the results for case 9 support the hypothesis that a hitherto unidentified SHOX enhancer resides within the ~500 kb region ~300 kb downstream of the exons.23, 24 Likewise, the results of case 16 are consistent with the recently proposed notion that the duplications in the downstream region of the known CNEs can underlie ISS and LWD.25 The high frequency and heterogeneity of CNVs in cases 1–16 may reflect the genomic instability of PAR1; the recombination rate of PAR1 during male meiosis is ~17-times higher than the genomic average.26 Indeed, the breakpoints of cases 2, 3, 5, 12 and 14 were located close to or within repetitive sequences, which provide substrates for non-allelic homologous recombination.27 While the breakpoints of cases 1–16 were widely distributed in PAR1 and relatively frequent in the genomic regions adjacent to SHOX, this is consistent with the occurrence of male-specific crossover throughout PAR1 with hotspots in the ~0.8 Mb genomic interval around SHOX.26
Probably damaging intragenic mutations were detected in cases 17–20. These data confirm the results of the European studies that the pathogenic point mutations in SHOX are widely distributed in the coding region and relatively common in the homeobox.2, 28 On the other hand, the pathogenicity of four benign/silent substitutions in SHOX exons and ten substitutions in the CNEs remains unknown. As three of the ten substitutions in the CNEs resided within putative enhancer sequences, these substitutions may disturb SHOX expression. However, most of the affected nucleotides were not highly conserved among species, raising a question of the functional significance of the substitutions. Furthermore, most of these substitutions were shared by control individuals. Our data imply that the point mutations in the known CNEs account for only a minor fraction of the etiology of SHOX haploinsufficiency, if any. As additional SHOX enhancers are likely to reside in PAR1,23, 24 sequence analysis of these novel enhancer regions is needed.
In summary, our results indicate that the frequency and mutation spectrum of SHOX abnormalities are comparable between Asian and European patients, with the exception of a European-specific downstream deletion. Furthermore, this study highlights the clinical significance and genetic heterogeneity of PAR1-linked CNVs, and implies a limited role of CNE-linked mutations in the etiology of ISS and LWD.
References
Rao, E., Weiss, B., Fukami, M., Rump, A., Niesler, B., Mertz, A. et al. Pseudoautosomal deletions encompassing a novel homeobox gene cause growth failure in idiopathic short stature and Turner syndrome. Nat. Genet. 16, 54–63 (1997).
Binder, G. Short stature due to SHOX deficiency: genotype, phenotype, and therapy. Horm. Res. Paediatr. 75, 81–89 (2011).
Seki, A., Jinno, T., Suzuki, E., Takayama, S., Ogata, T. & Fukami, M. Skeletal deformity associated with SHOX deficiency. Clin. Pediatr. Endocrinol 23, 65–72 (2014).
Huber, C., Cusin, V., Le Merrer, M., Mathieu, M., Sulmont, V., Dagoneau, N. et al. SHOX point mutations in dyschondrosteosis. J. Med. Genet. 38, 323 (2001).
Ross, J. L., Scott, C. Jr., Marttila, P., Kowal, K., Nass, A., Papenhausen, P. et al. Phenotypes associated with SHOX deficiency. J. Clin. Endocrinol. Metab. 86, 5674–5680 (2001).
Rappold, G. A., Fukami, M., Niesler, B., Schiller, S., Zumkeller, W., Bettendorf, M. et al. Deletions of the homeobox gene SHOX (short stature homeobox) are an important cause of growth failure in children with short stature. J. Clin. Endocrinol. Metab. 87, 1402–1406 (2002).
Benito-Sanz, S., Barroso, E., Heine-Suner, D., Hisado-Oliva, A., Romanelli, V., Rosell, J. et al. Clinical and molecular evaluation of SHOX/PAR1 duplications in Leri–Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J. Clin. Endocrinol. Metab. 96, E404–E412 (2011).
Benito-Sanz, S., Thomas, N. S., Huber, C., Gorbenko del Blanco, D., Aza-Carmona, M., Crolla, J. A. et al. A novel class of Pseudoautosomal region 1 deletions downstream of SHOX is associated with Leri–Weill dyschondrosteosis. Am. J. Hum. Genet. 77, 533–544 (2005).
Benito-Sanz, S., Royo, J. L., Barroso, E., Paumard-Hernandez, B., Barreda-Bonis, A. C., Liu, P. et al. Identification of the first recurrent PAR1 deletion in Leri–Weill dyschondrosteosis and idiopathic short stature reveals the presence of a novel SHOX enhancer. J. Med. Genet. 49, 442–450 (2012).
Benito-Sanz, S., Aza-Carmona, M., Rodriguez-Estevez, A., Rica-Etxebarria, I., Gracia, R., Campos-Barros, A. et al. Identification of the first PAR1 deletion encompassing upstream SHOX enhancers in a family with idiopathic short stature. Eur. J. Hum. Genet. 20, 125–127 (2012).
Fukami, M., Kato, F., Tajima, T., Yokoya, S. & Ogata, T. Transactivation function of an approximately 800-bp evolutionarily conserved sequence at the SHOX 3′ region: implication for the downstream enhancer. Am. J. Hum. Genet. 78, 167–170 (2006).
Chen, J., Wildhardt, G., Zhong, Z., Roth, R., Weiss, B., Steinberger, D. et al. Enhancer deletions of the SHOX gene as a frequent cause of short stature: the essential role of a 250 kb downstream regulatory domain. J. Med. Genet. 46, 834–839 (2009).
Durand, C., Bangs, F., Signolet, J., Decker, E., Tickle, C. & Rappold, G. Enhancer elements upstream of the SHOX gene are active in the developing limb. Eur. J. Hum. Genet. 18, 527–532 (2010).
Sabherwal, N., Bangs, F., Roth, R., Weiss, B., Jantz, K., Tiecke, E. et al. Long-range conserved non-coding SHOX sequences regulate expression in developing chicken limb and are associated with short stature phenotypes in human patients. Hum. Mol. Genet. 16, 210–222 (2007).
Verdin, H., Fernandez-Minan, A., Benito-Sanz, S., Janssens, S., Callewaert, B., Waele, K. D. et al. Profiling of conserved non-coding elements upstream of SHOX and functional characterisation of the SHOX cis-regulatory landscape. Sci. Rep. 5, 17667 (2015).
Huber, C., Rosilio, M., Munnich, A. & Cormier-Daire, V. High incidence of SHOX anomalies in individuals with short stature. J. Med. Genet. 43, 735–739 (2006).
Rappold, G., Blum, W. F., Shavrikova, E. P., Crowe, B. J., Roeth, R., Quigley, C. A. et al. Genotypes and phenotypes in children with short stature: clinical indicators of SHOX haploinsufficiency. J. Med. Genet. 44, 306–313 (2007).
Hirschfeldova, K., Solc, R., Baxova, A., Zapletalova, J., Kebrdlova, V., Gaillyova, R. et al. SHOX gene defects and selected dysmorphic signs in patients of idiopathic short stature and Leri–Weill dyschondrosteosis. Gene 491, 123–127 (2012).
Bunyan, D. J., Baker, K. R., Harvey, J. F. & Thomas, N. S. Diagnostic screening identifies a wide range of mutations involving the SHOX gene, including a common 47.5 kb deletion 160 kb downstream with a variable phenotypic effect. Am. J. Med. Genet. Part A 161a, 1329–1338 (2013).
Kant, S. G., Broekman, S. J., de Wit, C. C., Bos, M., Scheltinga, S. A., Bakker, E. et al. Phenotypic characterization of patients with deletions in the 3'-flanking SHOX region. PeerJ 1, e35 (2013).
Fukami, M., Naiki, Y., Muroya, K., Hamajima, T., Soneda, S., Horikawa, R. et al. Rare pseudoautosomal copy-number variations involving SHOX and/or its flanking regions in individuals with and without short stature. J. Hum. Genet. 60, 553–556 (2015).
Hisado-Oliva, A., Garre-Vazquez, A. I., Santaolalla-Caballero, F., Belinchon, A., Barreda-Bonis, A. C., Vasques, G. A. et al. Heterozygous NPR2 mutations cause disproportionate short stature, similar to Leri–Weill dyschondrosteosis. J. Clin. Endocrinol. Metab. 100, E1133–E1142 (2015).
Bunyan, D. J., Taylor, E. J., Maloney, V. K. & Blyth, M. Homozygosity for a novel deletion downstream of the SHOX gene provides evidence for an additional long range regulatory region with a mild phenotypic effect. Am. J. Med. Genet. Part A 164a, 2764–2768 (2014).
Tsuchiya, T., Shibata, M., Numabe, H., Jinno, T., Nakabayashi, K., Nishimura, G. et al. Compound heterozygous deletions in pseudoautosomal region 1 in an infant with mild manifestations of langer mesomelic dysplasia. Am. J. Med. Genet. Part A 164a, 505–510 (2014).
Bunyan, D. J., Baffico, M., Capone, L., Vannelli, S., Iughetti, L., Schmitt, S. et al. Duplications upstream and downstream of SHOX identified as novel causes of Leri–Weill dyschondrosteosis or idiopathic short stature. Am. J. Med. Genet. Part A:, (e-pub ahead of print 24 December 2015; doi:10.1002/ajmg.a.3752).
Hinch, A. G., Altemose, N., Noor, N., Donnelly, P. & Myers, S. R. Recombination in the human Pseudoautosomal region PAR1. PLoS Genet. 10, e1004503 (2014).
Gu, W., Zhang, F. & Lupski, J. R. Mechanisms for human genomic rearrangements. PathoGenetics 1, 4 (2008).
Marchini, A., Rappold, G. & Schneider, K. U. SHOX at a glance: from gene to protein. Arch. Physiol. Biochem. 113, 116–123 (2007).
Acknowledgements
We thank Drs M Toki and M Izawa for providing samples and clinical information of their patients. This study was supported by JCR Pharmaceuticals. The sponsor had no role in the study design, in the collection, analysis or interpretation of data, in the writing of the report or in the decision to submit the report for publication.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
Maki Fukami has received a research grant from JCR pharmaceuticals. The remaining authors declare no conflict of interest.
Additional information
The Japanese SHOX Study Group M. Adachi (Kanagawa Children’s Medical Center), T. Tajima (Hokkaido University), T. Tanaka (Tanaka Growth Clinic), O. Arisaka and S. Koyama (Dokkyo Medical University), T. Hamajima (Aichi Children’s Health and Medical Center), O. Nose (Nose Clinic), K. Ozono and N. Namba (Osaka University), K. Nagasaki (Niigata University), T. Kamimaki (Shizuoka City Shimizu Hospital), S. Kanzaki (Tottori University), T. Ogata (Hamamatsu University School of Medicine), H. Tanaka (Okayama Saiseikai General Hospital), Y. Hasegawa (Tokyo Metropolitan Children’s Medical Center), K. Kobayashi (University of Yamanashi), S. Dateki (Nagasaki University), H. Mabe (Kumamoto University), I. Fujiwara (Tohoku University), S. Ida (Osaka Medical Center and Research Institute for Maternal and Child Health), T. Hasegawa (Keio University), A. Uematsu (Shizuoka Children’s Hospital), K. Kashimada (Tokyo Medical Dental University), K. Onigata (Shimane University), K. Miyako (Fukuoka Children’s Hospital), S. Yokoya and R. Horikawa (National Center for Child Health and Development), and M. Fukami (National Research Institute for Child Health and Development).
Supplementary Information accompanies the paper on Journal of Human Genetics website .
Rights and permissions
About this article

Cite this article
Shima, H., Tanaka, T., Kamimaki, T. et al. Systematic molecular analyses of SHOX in Japanese patients with idiopathic short stature and Leri–Weill dyschondrosteosis. J Hum Genet 61, 585–591 (2016). https://doi.org/10.1038/jhg.2016.18
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2016.18
This article is cited by
-
Rare dosage abnormalities flanking the SHOX gene
Egyptian Journal of Medical Human Genetics (2021)
-
Novel aggrecan variant, p. Gln2364Pro, causes severe familial nonsyndromic adult short stature and poor growth hormone response in Chinese children
BMC Medical Genetics (2018)
-
Identification of a novel heterozygous mutation of the Aggrecan gene in a family with idiopathic short stature and multiple intervertebral disc herniation
Journal of Human Genetics (2017)
