
Abstract
There is an overlap between the physiological cardiac remodeling associated with training in athletes, the so-called athlete’s heart, and mild forms of hypertrophic cardiomyopathy (HCM), the most common hereditary cardiac disease. HCM is often accompanied by unfavorable outcomes including a sudden cardiac death in the adolescents. Because one of the initial signs of HCM is abnormality in electrocardiogram (ECG), athletes may need to monitor for ECG findings to prevent any unfavorable outcomes. HCM is caused by mutations in genes for sarcomere proteins, but there is no report on the systematic screening of gene mutations in athletes. One hundred and two genetically unrelated young Japanese athletes with abnormal ECG findings were the subjects for the analysis of four sarcomere genes, MYH7, MYBPC3, TNNT2 and TNNI3. We found that 5 out of 102 (4.9%) athletes carried mutations: a heterozygous MYH7 Glu935Lys mutation, a heterozygous MYBPC3 Arg160Trp mutation and another heterozygous MYBPC3 Thr1046Met mutation, all of which had been reported as HCM-associated mutations, in 1, 2 and 2 subjects, respectively. This is the first study of systematic screening of sarcomere gene mutations in a cohort of athletes with abnormal ECG, demonstrating the presence of sarcomere gene mutations in the athlete’s heart.
Similar content being viewed by others


Introduction
Cardiac hypertrophy is an adaptive response of the heart, in which myocardial mass is increased beyond the normal range. Various causes could lead to cardiac hypertrophy, but in general it occurs in response to hemodynamic (volume or pressure) overload.1 Although the most common etiologies of pathological cardiac hypertrophy are high blood pressure (hypertension) and valvular heart diseases, intensive endurance or strength training may result in physiological cardiac hypertrophy, called as ‘athlete’s heart’. Intensive prolonged exercise increases the aerobic capacity and oxygen consumption of skeletal muscles, followed by increases of diastolic ventricular filling and stroke volume, leading to a rise in maximum cardiac output and peak oxygen consumption.2 Induced volume overload increases not only end-diastolic and end-systolic ventricular volumes but also the corresponding diameters. Consequently, compensatory myocardial hypertrophy develops without external stimuli.2 The physiological left ventricular (LV) remodeling occasionally mimics certain pathological conditions associated with sudden cardiac death (SCD), such as hypertrophic cardiomyopathy (HCM).2, 3 Indeed, it is often difficult to distinguish the ‘athlete’s heart’ from HCM, because there are overlaps in phenotypes between ‘athlete’s heart’ and mild form of HCM.2, 4 It was reported that HCM had consistently been the most common cause of SCD in young athletes.3 Therefore, early differential diagnosis of HCM from the physiological cardiac hypertrophy is important to prevent the unfavorable outcome in the athletes.
Participation in intense competitive sports can represent a potential risk factor of SCD in athletes with HCM, even when conventional markers are absent.5 It was strongly suggested that the diagnostic ambiguity should be solved to reduce the risk for SCD, by using the paradigm of noninvasive parameters including findings in electrocardiogram (ECG), echocardiogram and genetic analysis of HCM-causing sarcomere gene mutations.4, 6, 7 It has been reported that ECG is useful to detect early signs of HCM, but there is no report on the systematic screening of sarcomere gene mutations in a panel of trained athletes.8 In this study, we analyzed four major HCM-causing sarcomere genes, MYH7, MYBPC3, TNNT2 and TNNI3,9 in 102 unrelated young Japanese athletes to clarify the genetic predisposing factors.
Materials and methods
Subjects
A total of 102 genetically unrelated Japanese trained athletes between the ages of 18 and 28 were the subjects in this study. None of the athletes in this study had apparent family history of HCM or SCD in first-degree relatives, but they had shown abnormal ECG changes or mild cardiomegaly found in a chest X-ray examination. ECG abnormalities include LV hypertrophy, ST-T changes and/or conduction block determined by the criteria as reported previously,8 in which signs of LV hypertrophy in ECG were observed in 62 subjects. Echocardiographic-based LV hypertrophy was noted in only 7 out of the 102 athletes. In addition, genotyping data from our HCM cohort composing of 162 familial cases and 100 sporadic cases10 were retrieved for specific mutations and previously obtained samples from patients with specific mutations and their family relatives were analyzed.
Written informed consent was obtained from each subject and the research protocol conformed to the ethical guidelines of the Declaration of Helsinki as reflected in a priori approval by the Ethics Committees of Juntendo University School of Health and Sports Science and Medical Research Institute of Tokyo Medical and Dental University.
Genetic analyses
DNA samples extracted from peripheral blood of subjects were used as templates to amplify each coding exon of MYBPC3 (NM_000256), TNNT2 (NM_001001430) and TNNI3 (NM_000363), and exons 3–25 of MYH7 (MN_000257) by PCR and the PCR products were analyzed for sequence variations by direct DNA sequencing on both strands by using Big Dye Terminator (version 3.1) Cycle Sequencing kit and ABI3130xl DNA Analyzer (Applied Biosystems, Foster City, CA, USA), as described previously.9
Haplotype analyses of sarcomere gene mutations
Microsatellite analyses were done for MYOI with primers of 5′-NED-CAGGTCTAAACATGGGCG-3′ and 5′-ACATACCCTTTCTCACATTCAG-3′ and MYOII with primers of 5′VIC-GTGAGTAGATTGAGAGTTGTGGG-3′ and 5′-TCCTCTAACCCTACCCCCC-3′ in MYH7 locus11 and MYBPC3-CA with primers of 5′-FAM-GGTCAGGTCACCTAGCATAGCAT-3′ and 5′-GGCAGATTCCTGATTTATTTGGC-3′ and D11S4109 with primers of 5′-PET-AGTTAGGAGACCTGGCATTTCTC-3′ and 5′-GAAGATCCCTCACAGACTCTCTCT-3′ in MYBPC3 locus.12 PCR fragments amplified with the primers were electrophoresed in the ABI3130xl DNA sequencer (Applied Biosystems) and the microsatellite alleles were defined as length of PCR fragments. The microsatellite polymorphisms were confirmed by sequencing PCR fragments amplified with nonlabeled primers.
In silico prediction of functional impacts
Pathogenicity of the identified mutation was predicted by using online programs, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Taster (http://www.mutationtaster.org/) and PROVEN (http://provean.jcvi.org/index.php). We searched for data concerning pathogenicity of mutations in the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/).
Results
Identification of sarcomere gene mutations in trained athletes
To explore the possible presence of HCM-causing mutations in Japanese athletes, 102 trained athletes were tested for mutations in four sarcomere genes—MYH7, MYBPC3, TNNT2 and TNNI3—because these four genes are the major HCM-causing genes in Japan.9, 10 In addition to 10 synonymous polymorphisms (rs2069540, rs2069542, rs735712, rs2231126 and rs7157716 in MYH7, rs11570058, rs379953 and rs1052373 in MYBPC3, rs3729547 in TNNT2 and rs3729841 in TNNI3), four missense variations were identified (Table 1). Among the missense variations, Lys253Arg (c.758A>G, rs3730238) in TNNT2 was found in seven subjects and it was a common polymorphism registered in the dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/), the 1000 genomes database (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/) and the human genetic variation database (http://www.genome.med.kyoto-u.ac.jp/SnpDB/). The other three missense variants, Glu935Lys (c.2803G>A, rs121913639) in MYH7 found in one case (Jun-49), and Arg160Trp (c.478C>T, rs193068692) and Thr1046Met (c.3137C>T, rs371061770) in MYBPC3 found in two (Jun-3 and Jun-16) and two (Jun-75 and Jun-78) subjects, respectively, were previously reported as HCM-associated mutations.13, 14 The MYBPC3 Thr1046Met mutation was found at rare frequencies in healthy subjects in the 1000 genomes database and human genetic variation database, although the MYH7 Glu935Lys and MYBPC3 Arg160Trp mutations were not found in Japanese populations registered in the 1000 genomes database and human genetic variation database (Table 1).
Pathological impacts of sarcomere gene mutations and haplotype analysis
To investigate the possible pathological roles of the missense variants, their functional impacts were predicted by in silico analysis using PolyPhen-2, Mutation Taster and PROVEN. As shown in Table 1, MYBPC3 Thr1046Met was suggested to be a benign mutation or a polymorphism, as was the case with the TNNT2 Lys253Arg polymorphism. On the other hand, MYH7 Glu935Lys and MYBPC3 Arg160Trp were predicted to be disease-causing mutations. We, then, searched for these two mutations in our HCM cohort consisting of 162 familial and 100 sporadic cases,10 and found the MYH7 Glu935Lys in three (two familial and one sporadic) patients and the MYBPC3 Arg160Trp in two (one familial and one sporadic) patients. Because family samples were available from each one family for the MYH7 and MYBPC3 mutations, we investigate the linkage of these mutations with HCM in the multiplex families along with the analyses of microsatellite markers.
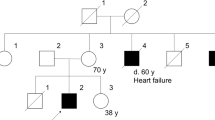
As shown in Figure 1, we found that the MYBPC3 Arg160Trp mutation was not co-segregated with HCM in a multiplex family with HCM originated from Tohoku area, because one subject showing LV hypertrophy and diastolic dysfunction did not carry the MYBPC3 mutation (Figure 1a). A microsatellite analysis of the MYBPC3 locus showed that the MYBPC3 mutation (160Trp allele) was linked to a haplotype composed of allele 314 of MYBPC3-CA and allele 158 of D11S4109 in this family (Figure 1a). We also analyzed the other HCM patient from Kyushu area and two athletes, both from Kanto area, who carried the MYBPC3 mutation, for the microsatellite markers. It was revealed that they also shared the 160Trp-314-158 haplotype, suggesting a common ancestral origin of the MYBPC3 mutation (Supplementary Table S1).

Linkage studies for MYBPC3 and MYH7 mutations in multiplex families with HCM. (a) Pedigree of a HCM family with the MYBPC3 Arg160Trp mutation. MYBPC3 haplotypes composed of rs193068692 (Arg160Trp)-MYBPC3-CA (fragment length)-D11S4109 (fragment length) were deduced from the genotyping data and indicated with ID of subjects. (b) Pedigree of a HCM family with the MYH7 Glu935Lys mutation. MYH7 haplotypes consisting of MYOI (fragment length)-rs2069540 (C/T)-rs2069542 (C/T)-rs735712 (C/T)-rs2231126 (C/T)-rs121913639 (Glu935Lys)-rs7157716 (C/T)-MYOII (fragment length) were deduced from the genotyping data and indicated with ID of subjects. Owing to the phase ambiguity, haplotypes could not be determined in three subjects. As well, MYOI–MYOII haplotype could not be determined due to the phase ambiguity and indicated tentatively. Arrows indicate proband patients. dHCM, dilated phase HCM; HCM, hypertrophic cardiomyopathy; LVH, ECG findings of LV hypertrophy; NA, no abnormality in cardiac phenotype; SCD, sudden cardiac death.
On the other hand, a microsatellite analysis of the MYH7 locus in the other multiplex family from Kyushu area showed a linkage of MYH7 Glu935Lys with HCM (Figure 1b). Although the parents with the MYH7 Glu935Lys mutation were cousins,13 it was found that they carried different MYH7-mutation-linked haplotypes (Figure 1b), because both sons, who were homozygous for the MYH7 mutation, were heterozygous for both MYOI and MYOII loci (Figure 1b). This observation indicated that there were at least two different haplotypes harboring the MYH7 mutation. By analyzing the other two HCM patients from Kansai area and Kanto area and the athlete from Kanto area, who had the MYH7 mutation, we obtained the data consistent with that the MYOI (intron 1)-rs2069540 (exon3)-rs2069542 (exon 8)-rs735712 (exon 12)-rs2231126 (exon 12)-rs121913639 (exon 23, Glu935Lys)-rs7157716 (exon 24)-MYOII (intron 24) haplotype of 164-C-C-C-C-935Lys-T-148 was shared by all of them, implying the common ancestral origin of the MYH7 mutation (Supplementary Table S2).
Clinical phenotype of the athletes carrying the sarcomere gene mutations
The athlete carrying the MYH7 Glu935Lys mutation was a 19-year-old male who played football. His ECG showed high voltages (voltage of R wave in V5 lead: 33 mm, voltage of S wave in V1 lead: 26 mm) and q waves in II, III, aVF, V5, and V6 leads, indicating a sign of LV hypertrophy, although no LV hypertrophy was evident in his echocardiography. Phenotypes of the other four subjects carrying the MYBPC3 Arg160Trp or Thr1046Met variant are also shown in Table 2. Of note was that an athlete with the MYBPC Arg160Trp mutation was shown to have asymmetric cardiac hypertrophy in his echocardiography, which is a typical cardiac phenotype of HCM.
Discussion
In this study, three sarcomere gene mutations that were reported to be associated with HCM were identified in five trained athletes. It was reported that the prevalence of HCM in elite athletes was significantly less than that in the general population (1 in 500 in the general subjects3), suggesting that structural and functional changes associated with HCM might naturally select-out most individuals with HCM from competitive sports.15 However, a minority of HCM patients showed relatively mild LV hypertrophy and many of these patients are asymptomatic.16 Actually, a small but substantial subset of elite athletes manifested with a slight increase of LV wall thickness, which defined a gray zone of overlap between the extreme expressions of ‘athlete’s heart’ and a mild HCM form.4 In addition, the most common cause of SCD in young athletes appears to be HCM.16, 17 These observations suggest the critical importance to clarify whether the increased LV wall thickness would represent a physiological adaptation of heart to athletic training or a pathological condition such as HCM. This ambiguity needs to be solved by applying a number of noninvasive parameters including a genetic testing to prevent athletes from SCD, although the causative role of the sarcomere gene mutations should be carefully considered.
Prevalence of the mutations in nucleotide variations database from healthy individuals including dbSNP, 1000 genomes and human genetic variation databases, description of clinical significance in the ClinVar database, and in silico studies would be helpful to predict the pathological significance of identified mutations. In this study, MYBPC3 Thr1046Met was suggested to be benign and its clinical significance was debatable, although we could not exclude a possibility of its role in promoting cardiac hypertrophy, because the mutation had been reported in patients with elderly onset HCM who might have any confounding factors including mild hypertension.14 In addition, MYBPC3 Arg160Trp was not linked to HCM in a multiplex family tested in this study, indicating that it was not the cause of HCM for all the HCM patients in this family. However, a possibility of another disease-causing mutation in the MYBPC3 Arg160Trp-negative HCM patient could not be excluded and therefore MYBPC3 Arg160Trp might be a responsible mutation in the other HCM patients (Figure 1a), because in silico studies strongly suggested functional alterations by this rare variant.
On the other hand, MYH7 Glu935Lys was not found in the healthy individuals in dbSNP, 1000 genomes and human genetic variation databases, listed as pathogenic mutation in the ClinVar database, and predicted in silico to have functional impacts in this study. We previously reported the phenotypes of MYH7 Glu935Lys mutation in the Japanese HCM family (Figure 1b).13 In this family, the proband patient and his elder brother were homozygous for the mutation and showed dilated phase of HCM and severe LV hypertrophy, respectively, and both eventually died at their thirties. In contrast, their parents were heterozygous for the mutation and both showed mild LV hypertrophy in the presence of blood hypertension.13 Their elder sister was also heterozygous for the mutation, but she was normotensive and did not manifest with LV hypertrophy. These observations suggested a gene dose effect of the mutation on the clinical manifestation of HCM, that is, homozygotes were severer than the heterozygotes, as well as an age- or blood pressure-dependent penetrance of HCM caused by this mutation. Therefore, it can be speculated that the athlete carrying the MYH7 Glu935Lys mutation in the heterozygous state might develop apparent cardiac hypertrophy or HCM phenotypes in the future, if he would continue the athletic training or when he would develop hypertension. Continuous and careful follow-up will be required for him to evaluate the clinical phenotypes caused by the mutation and/or athletic training. Similarly, careful follow-up is required for two athletes with the MYBPC3 Arg160Trp mutation, especially for a subject who showed asymmetric cardiac hypertrophy in his echocardiography (Table 2).
In conclusion, we report here for the first time the results of a systematic screening for the HCM-associated sarcomere gene mutations in young athletes with abnormal ECG findings. We identified one and two missense mutations in MYH7 and MYBPC3, respectively, which were reported to be the HCM-associated mutations, in five athletes. Of note was that 4 of them showed an apparent sign of LV hypertrophy in ECG and such abnormality was noted in 62 of 102 athletes, indicating that the sarcomere gene mutations were found in 4 of 62 (6.5%) athletes with ECG sign of LV hypertrophy. This result indicates that the potential application of genetic testing in trained athletes provides a definitive diagnosis of HCM in subjects with early ECG signs of LV wall thickness. Further genetic analyses in the other HCM genes for sarcomere proteins will help understand the genetic factors in young athletes with difficult diagnostic situation or with borderline findings of HCM.
References
Olivetti, G., Cigola, E., Maestri, R., Lagrasta, C., Corradi, D. & Quaini, F. Recent advances in cardiac hypertrophy. Cardiovasc. Res. 45, 68–75 (2000).
Lauschke, J. & Maisch, B. Athlete’s heart or hypertrophic cardiomyopathy? Clin. Res. Cardiol. 98, 80–88 (2009).
Maron, B. J. Sudden death in young athletes. N. Engl. J. Med. 349, 1064–1075 (2003).
Pelliccia, A., Maron, M. S. & Maron, B. J. Assessment of left ventricular hypertrophy in a trained athlete: differential diagnosis of physiologic athlete’s heart from pathologic hypertrophy. Prog. Cardiovasc. Dis. 54, 387–396 (2012).
Maron, B. J. Risk stratification and role of implantable defibrillators for prevention of sudden death in patients with hypertrophic cardiomyopathy. Circ. J. 74, 2271–2282 (2010).
Fuller, C. M., McNulty, C. M., Spring, D.A., Arger, K. M., Bruce, S. S., Chryssos, B. E. et al. Prospective screening of 5615 high school athletes for risk of sudden cardiac death. Med. Sc. Sports Exerc. 29, 1131–1138 (1997).
Corrado, D., Basso, C., Schiavon, M. & Thiene, G. Screening for hypertrophic cardiomyopathy in young athletes. N. Engl. J. Med. 339, 364–369 (1998).
Pelliccia, A., Maron, B. J., Culasso, F., Di Paolo, F. M., Spataro, A., Biffi, A. et al. Clinical significance of abnormal electrocardiographic patterns in trained athletes. Circulation 102, 278–284 (2000).
Otsuka, H., Arimura, T., Abe, T., Kawai, H., Aizawa, Y., Kubo, T. et al. Prevalence and distribution of sarcomeric gene mutations in Japanese patients with familial hypertrophic cardiomyopathy. Circ. J. 76, 453–461 (2012).
Kimura, A. Molecular basis of hereditary cardiomyopathy: abnormalities in calcium sensitivity, stretch response, stress response and beyond. J. Hum. Genet. 55, 81–90 (2010).
Richard, P., Isnard, R., Carrier, L., Dubourg, O., Donatien, Y., Mathieu, B. et al. Double heterozygosity for mutations in the beta-myosin heavy chain and in the cardiac myosin binding protein C genes in a family with hypertrophic cardiomyopathy. J. Med. Genet. 36, 542–545 (1999).
Kubo, T., Kitaoka, H., Okawa, M., Matsumura, Y., Hitomi, N., Yamasaki, N. et al. Lifelong left ventricular remodeling of hypertrophic cardiomyopathy caused by a founder frameshift deletion mutation in the cardiac myosin-binding protein C gene among Japanese. J. Am. Coll. Cardiol. 46, 1737–1743 (2005).
Nishi, H., Kimura, A., Harada, H., Adachi, K., Koga, Y., Sasazuki, T. et al. Possible gene dose effect of a mutant cardiac beta-myosin heavy chain gene on the clinical expression of familial hypertrophic cardiomyopathy. Biochem. Biophys. Res. Commun. 200, 549–556 (1994).
Anan, R., Niimura, H., Takenaka, T., Hamasaki, S. & Tei, C. Mutations in the genes for sarcomeric proteins in Japanese patients with onset sporadic hypertrophic cardiomyopathy after age 40 years. Am. J. Cardiol. 99, 1750–1754 (2007).
Basavarajaiah, S., Wilson, M., Whyte, G., Shah, A., McKenna, W. & Sharma, S. Prevalence of hypertrophic cardiomyopathy in highly trained athletes: relevance to pre-participation screening. J. Am. Coll. Cardiol. 51, 1033–1039 (2008).
Maron, B. J., Pelliccia, A. & Spirito, P. Cardiac disease in young trained athletes. Insights into methods for distinguishing athlete’s heart from structural heart disease, with particular emphasis on hypertrophic cardiomyopathy. Circulation 91, 1596–1601 (1995).
Erdmann, J., Daehmlow, S., Wischke, S., Senyuva, M., Werner, U., Raible, J. et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin. Genet. 64, 339–349 (2003).
Acknowledgements
We thank Ms Yukiko Ueda and Nana Ohkubo for their technical assistance and clinicians for their contribution in blood sampling and clinical examination of HCM patients and family relatives. This work was supported in part by Grant-in-Aids from the Ministry of Education, Culture, Sports, Science and Technology, Japan (25293181, 25670172 and 26460407); a research grant for Idiopathic Cardiomyopathy from the Ministry of Health, Labor and Welfare, Japan; and a research grant from Association Française contre les Myopathies. The study was also supported by Joint Usage/Research Program of Medical Research Institute, Tokyo Medical and Dental University and a support for women researchers from the Tokyo Medical and Dental University.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website .
Supplementary information
Rights and permissions
About this article

Cite this article
Kadota, C., Arimura, T., Hayashi, T. et al. Screening of sarcomere gene mutations in young athletes with abnormal findings in electrocardiography: identification of a MYH7 mutation and MYBPC3 mutations. J Hum Genet 60, 641–645 (2015). https://doi.org/10.1038/jhg.2015.81
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2015.81
This article is cited by
-
MYH7 in cardiomyopathy and skeletal muscle myopathy
Molecular and Cellular Biochemistry (2023)
