
Abstract
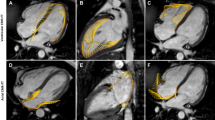
Whereas truncating variants of the giant protein Titin (TTNtv) are the main cause of familial dilated cardiomyopathy (DCM), recently Filamin C truncating variants (FLNCtv) were identified as a cause of arrhythmogenic cardiomyopathy (ACM). Our aim was to characterize and compare clinical and MRI features of TTNtv and FLNCtv in the Belgian population. In index patients referred for genetic testing of ACM/DCM, FLNCtv and TTNtv were found in 17 (3.6%) and 33 (12.3%) subjects, respectively. Further family cascade screening yielded 24 and 19 additional truncating variant carriers in FLNC and TTN, respectively. The main phenotype was ACM in FLNCtv carriers whereas TTNtv carriers showed either an ACM or DCM phenotype. Non-sustained Ventricular Tachycardia was frequent in both populations. MRI data, available in 28/40 FLNCtv and 32/52 TTNtv patients, showed lower Left Ventricular (LV) ejection fraction and lower LV strain in TTNtv patients (p < 0.01). Conversely, both the frequency (68% vs 22%) and extent of non-ischemic myocardial late gadolinium enhancement (LGE) was significantly higher in FLNCtv patients (p < 0.01). Hereby, ring-like LGE was found in 16/19 (84%) FLNCtv versus 1/7 (14%) of TTNtv patients (p < 0.01). In conclusion, a large number of FLNCtv and TTNtv patients present with an ACM phenotype but can be separated by cardiac MRI. Whereas FLNCtv patients often have extensive myocardial fibrosis, typically following a ring-like pattern, LV dysfunction without or limited replacement fibrosis is the common TTNtv phenotype.

This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
Data availability
The data underlying this article will be shared on reasonable request to the corresponding author. The genetic variants were deposited in ClinVar (accession numbers SCV002581936–SCV002581950 and SCV002817388–SCV002817419).
References
Garcia-Pavia P, Kim Y, Restrepo-Cordoba MA, Lunde IG, Wakimoto H, Smith AM, et al. Genetic variants associated with cancer therapy-induced cardiomyopathy. Circulation. 2019;140:31–41.
Henkens MTHM, Stroeks SLVM, Raafs AG, Sikking MA, Tromp J, Ouwerkerk W, et al. Dynamic ejection fraction trajectory in patients with dilated cardiomyopathy with a truncating titin variant. Circ Heart Fail. 2022;15:e009352.
Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–28.
Eldemire R, Tharp CA, Taylor MRG, Sbaizero O, Mestroni L. The sarcomeric spring protein titin: biophysical properties, molecular mechanisms, and genetic mutations associated with heart failure and cardiomyopathy. Curr Cardiol Rep. 2021;23:121.
Ware JS, Cook SA. Role of titin in cardiomyopathy: from DNA variants to patient stratification. Nat Rev Cardiol. 2018;15:241–52.
Akhtar MM, Lorenzini M, Cicerchia M, Ochoa JP, Hey TM, Sabater Molina M, et al. Clinical phenotypes and prognosis of dilated cardiomyopathy caused by truncating variants in the TTN gene. Circ Heart Fail. 2020;13:e006832.
Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, et al. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra6.
McAfee Q, Chen CY, Yang Y, Caporizzo MA, Morley M, Babu A, et al. Truncated titin proteins in dilated cardiomyopathy. Sci Transl Med. 2021;13:eabd7287.
Haggerty CM, Damrauer SM, Levin MG, Birtwell D, Carey DJ, Golden AM, et al. Genomics-first evaluation of heart disease associated with titin-truncating variants. Circulation. 2019;140:42–54.
Celeghin R, Cipriani A, Bariani R, Bueno Marinas M, Cason M, Bevilacqua M, et al. Filamin-C variant-associated cardiomyopathy: a pooled analysis of individual patient data to evaluate the clinical profile and risk of sudden cardiac death. Heart Rhythm. 2021;19:235–43.
Akhtar MM, Lorenzini M, Pavlou M, Ochoa JP, O’Mahony C, Restrepo-Cordoba MA, et al. Association of left ventricular systolic dysfunction among carriers of truncating variants in filamin C with frequent ventricular arrhythmia and end-stage heart failure. JAMA Cardiol. 2021;6:891–901.
Miles C, Finocchiaro G, Papadakis M, Gray B, Westaby J, Ensam B, et al. Sudden death and left ventricular involvement in arrhythmogenic cardiomyopathy. Circulation. 2019;139:1786–97.
Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, et al. Truncating FLNC mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. J Am Coll Cardiol. 2016;68:2440–51.
Verdonschot JAJ, Vanhoutte EK, Claes GRF, Helderman-van den Enden ATJM, Hoeijmakers JGJ, Hellebrekers DMEI, et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum Mutat. 2020;41:1091–111.
Gigli M, Stolfo D, Graw SL, Merlo M, Gregorio C, Nee Chen S, et al. Phenotypic expression, natural history, and risk stratification of cardiomyopathy caused by filamin C truncating variants. Circulation. 2021;144:1600–11.
Zegard A, Okafor O, de Bono J, Kalla M, Lencioni M, Marshall H, et al. Myocardial fibrosis as a predictor of sudden death in patients with coronary artery disease. J Am Coll Cardiol. 2021;77:29–41.
Leyva F, Zegard A, Okafor O, Foley P, Umar F, Taylor RJ, et al. Myocardial fibrosis predicts ventricular arrhythmias and sudden death after cardiac electronic device implantation. J Am Coll Cardiol. 2022;79:665–78.
Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, et al. HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart Rhythm. 2019;16:e373–407.
Zeppenfeld K, Tfelt-Hansen J, de Riva M, Winkel BG, Behr ER, Blom NA, et al. ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death. Eur Heart J. 2022;43:3997–4126.
Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, Coto E, Astudillo A, Puente DA, et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun. 2014;5:5326.
Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. Eur Heart J Cardiovasc Imaging. 2015;16:233–70.
Vandenberk B, Vandael E, Robyns T, Vandenberghe J, Garweg C, Foulon V, et al. Which QT correction formulae to use for QT monitoring? J Am Heart Assoc. 2016;5:1–10.
Augusto JB, Eiros R, Nakou E, Moura-Ferreira S, Treibel TA, Captur G, et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. Eur Heart J Cardiovasc Imaging. 2020;21:326–36.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Haverfield EV, Esplin ED, Aguilar SJ, Hatchell KE, Ormond KE, Hanson-Kahn A, et al. Physician-directed genetic screening to evaluate personal risk for medically actionable disorders: a large multi-center cohort study. BMC Med. 2021;19:199.
Ritter A, Bedoukian E, Berger JH, Copenheaver D, Gray C, Krantz I, et al. Clinical utility of exome sequencing in infantile heart failure. Genet Med. 2020;22:423–6.
Lee S-H, Oh J, Lee S-T, Won D, Kim S, Choi H-K, et al. Generation of a human induced pluripotent stem cell line YCMi004-A from a patient with dilated cardiomyopathy carrying a protein-truncating mutation of the Titin gene and its differentiation towards cardiomyocytes. Stem Cell Research. 2022;59:102629.
Töpf A, Johnson K, Bates A, Phillips L, Chao KR, England EM, et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genet Med. 2020;22:1478–88.
Vissing CR, Rasmussen TB, Dybro AM, Olesen MS, Pedersen LN, Jensen M, et al. Dilated cardiomyopathy caused by truncating titin variants: long-term outcomes, arrhythmias, response to treatment and sex differences. J Med Genet. 2021;58:832–41.
Choi SH, Weng LC, Roselli C, Lin H, Haggerty CM, Shoemaker MB, et al. Association between titin loss-of-function variants and early-onset atrial fibrillation. JAMA. 2018;320:2354–64.
Cambon-Viala M, Gerard H, Nguyen K, Richard P, Ader F, Pruny JF, et al. Phenotype/genotype relationship in left ventricular noncompaction: ion channel gene mutations are associated with preserved left ventricular systolic function and biventricular noncompaction: phenotype/genotype of noncompaction. J Card Fail. 2021;27:677–81.
Koh HY, Haghighi A, Keywan C, Alexandrescu S, Plews-Ogan E, Haas EA, et al. Genetic determinants of sudden unexpected death in pediatrics. Genet Med. 2022;24:839–50.
Te Rijdt WP, Ten Sande JN, Gorter TM, van der Zwaag PA, van Rijsingen IA, Boekholdt SM, et al. Myocardial fibrosis as an early feature in phospholamban p.Arg14del mutation carriers: phenotypic insights from cardiovascular magnetic resonance imaging. Eur Heart J Cardiovasc Imaging. 2019;20:92–100.
Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC, et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation. 2020;141:1872–84.
Segura-Rodríguez D, Bermúdez-Jiménez FJ, Carriel V, López-Fernández S, González-Molina M, Oyonarte Ramírez JM, et al. Myocardial fibrosis in arrhythmogenic cardiomyopathy: a genotype-phenotype correlation study. Eur Heart J Cardiovasc Imaging. 2020;21:378–86.
Wang W, Murray B, Tichnell C, Gilotra NA, Zimmerman SL, Gasperetti A, et al. Clinical characteristics and risk stratification of desmoplakin cardiomyopathy. Europace. 2022;24:268–77.
Zghaib T, Te Riele ASJM, James CA, Rastegar N, Murray B, Tichnell C, et al. Left ventricular fibro-fatty replacement in arrhythmogenic right ventricular dysplasia/cardiomyopathy: prevalence, patterns, and association with arrhythmias. J Cardiovasc Magn Reson. 2021;23:58.
Pilichou K, Lazzarini E, Rigato I, Celeghin R, De Bortoli M, Perazzolo Marra M, et al. Large genomic rearrangements of desmosomal genes in Italian arrhythmogenic cardiomyopathy patients. Circ Arrhythm Electrophysiol. 2017;10:1–14.
Harper AR, Goel A, Grace C, Thomson KL, Petersen SE, Xu X, et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet. 2021;53:135–42.
Verdonschot JAJ, Hazebroek MR, Derks KWJ, Barandiarán Aizpurua A, Merken JJ, Wang P, et al. Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long-term life-threatening arrhythmias. Eur Heart J. 2018;39:864–73.
Savarese M, Vihola A, Oates EC, Barresi R, Fiorillo C, Tasca G, et al. Genotype-phenotype correlations in recessive titinopathies. Genet Med. 2020;22:2029–40.
Funding
RW is supported as postdoctoral clinical researcher by the Fund for Scientific Research Flanders. JB is supported by a senior clinical investigator fellowship by the Fund for Scientific Research Flanders.
Author information
Authors and Affiliations
Contributions
TR, JJ and JGB contributed to the study design. LVA, MD, WH, KDV, RW, JVC, JGB and TR contributed to data collection. JB, AC, CK and TR contributed to genetic analysis and interpretation. JGB analyzed and interpreted the MRI data. JJ, TR and JGB contributed to the database construction. JJ, TR and JGB analyzed the data and wrote the initial manuscript. JJ, TR and JGB performed additional analysis for the revision and wrote the revised manuscript. JJ designed the tables and figures. JJ, LVA, JB, AC, CK, MD, WH, KDV, RW, JVC, JGB and TR contributed to critical interpretation of the results and wrote the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
S66244 – ethical committee University Hospitals Leuven.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jacobs, J., Van Aelst, L., Breckpot, J. et al. Tools to differentiate between Filamin C and Titin truncating variant carriers: value of MRI. Eur J Hum Genet 31, 1323–1332 (2023). https://doi.org/10.1038/s41431-023-01357-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01357-1
This article is cited by
-
The next step toward personalized recommendations for genetic cardiomyopathies
European Journal of Human Genetics (2023)
-
Deep phenotyping and population-level data can help resolve genomic variants
European Journal of Human Genetics (2023)
