
Abstract
Classically, β-lactams need an ionizable group to potentiate antibacterial activity. Sets of cephalosporins and penicillins featuring different substituted hydroxamates in place of the traditional carboxylate group have been synthesized and tested for antibiotic activity. Many of the compounds exhibited anti-bacterial activities with notable MIC values in the range of 6–0.2 μM.
Similar content being viewed by others

Introduction
Among the various classes of antibiotics, the β-lactams continue to stand out as a hallmark of human accomplishment. This broad antibiotic group has saved countless numbers of lives and still makes up a majority of the antibiotic market today. The penicillins and cephalosporins represent the first two β-lactam scaffolds used clinically. These cores require an ionizable group to potentiate activity. For bicyclic β-lactams (for example, penams, carbapenams, cephalosporins), the ionizable group is a carboxylate, while the monocyclic β-lactams (monobactams) typically utilize a sulfonate (−SO3) on the β-lactam ring nitrogen (Figure 1).1, 2 Herein we report the incorporation of hydroxamates (pKa 9–10)3 or a hydroxamic acid (pKa ~6–7)3 at the ionizable position of representative penicillins and cephalosporins to determine whether they could act as alternate ionizable groups and promote antibacterial activity.

Classic β-lactam scaffolds and proposed hydroxamate alternative.
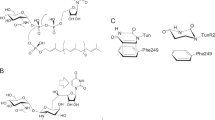
Over the last 80 years, a substantial number of person-years have gone into modifying the peripheral functionality around the β-lactam core. These modifications often involved variations of the penicillin C-6 and cephalosporin C-7 acylamino sidechains and especially functionality at the C-3′ position of cephalosporins (see Figure 1 for numbering). However, few reports have focused on modifying the ionizable position of penicillins and cephalosporins. Reports of penicillins and cephalosporins bearing hydroxamate or hydroxamic acid functionalities are very scarce in the literature. Brown4 reported the syntheses of penicillin G and ampicillin derivatives bearing amide and hydroxamic acid functionality in place of the traditional carboxylate, with several of the compounds exhibiting notable activity against Gram-positive bacteria. Further, Godfrey5 reported on the syntheses of a library of penicillin hydroxamates. Some of these reported compounds are illustrated in Figure 2. However, only select activities of these compounds were reported, with no broad antibacterial assessments and comparisons included.

Sample pencillins previously reported.
Related variations of cephalosporins included a library of 7-phenylacetyl and 7-thiophene-2-acetylamino cephalosporins bearing various carboxamides patented by Lewis.6 A number of these compounds, shown in Figure 3, were quite active, especially against S. aureus and B. cereus, exhibiting MIC values in the range of 6.25–0.003 μg ml−1. However, again this report did not provide broad antibacterial screenings and comparisons to classic penicillin and cephalosporin scaffolds. With these reports in mind, we were interested in expanding upon the studies of β-lactams bearing different hydroxamate functionality at the ionizable position. Additionally, broad biological assessments and structure activity relationships among the different series are also presented, including consequences of removal of the acidic hydroxamate proton by alkylation.

Sample cephalosporins previous reported.
Results and discussion
Our initial syntheses of cephalosporins are illustrated in Scheme 1. Commercially available 7-ACA was first acylated with phenylacetyl chloride to give 1, which was then coupled to both benzyl and O-TBS hydroxylamine to give 2a–b in good yields. Since removal of the benzyl group would likely be challenging in the presence of the divalent sulfur,7 deprotection of intermediate 2a to give 3 was not studied. Rather, intermediate 2b was deprotected with TBAF/AcOH to give 3 in 42% yield.
Efforts then focused on the syntheses of cephalosporins that featured the more complex 2-(2-amino-4-thiazolyl)-2(Z)-(methoxyimino)acetamido (ATMO)8 side chain that is often associated with a broader spectrum of antibiotic activity.8 Thus, cefotaxime sodium was coupled to both O-benzylhydroxylamine hydrochloride (OBHA•HCl) and O-allylhydroxylamine hydrochloride (OAHA•HCl) to give cephalosporins 4a–b (Scheme 2).
Attention then shifted to the syntheses of cephalosporins in which the hydroxamate proton was replaced with a methyl group. Syntheses of the necessary N-methyl-O-alkyl hydroxylamines9, 10 and their coupling to cephalosporins are illustrated in Schemes 3 and 4. Starting with either OBHA•HCl or OAHA•HCl (5a–b), reaction with Boc anhydride, followed by reaction with NaH, then methyl iodide gave intermediates 6a–b in good yields. Removal of the Boc group was accomplished with concentrated HCl to give desired salts 7a–b that were then neutralized and coupled to both cefotaxime sodium and cephalosporin 1 to give compounds 8–10.
Penicillin carboxylates were also converted to the corresponding hydroxamates. Attempted EDC-mediated coupling of O-tert-butyl(dimethylsilyl)hydroxylamine to cloxicillin sodium under organic conditions (Scheme 5) produced the desired product (11) in low yield with the major product being the rearranged N-acyl urea (12), produced during carbodiimide activation of the carboxyl group. We hypothesized that the combination of both a sterically bulky hydroxylamine and steric hindrance from the C-2 methyl groups of cloxicillin allowed the active O-acylisourea intermediate to rearrange faster than it was attacked by the hydroxylamine. A less sterically bulky hydroxylamine, O-allylhydroxylamine hydrochloride (OAHA•HCl), was employed and the coupling reaction was run under aqueous buffered conditions. As shown in Scheme 6, this change worked well and penams 13–14 were obtained in improved yield (Scheme 6). Next, penicillins in which the free hydroxamate proton was replaced with a methyl group were made (Scheme 7). Thus, N-Methyl-O-allyl hydroxylamine hydrochloride (7a, from Scheme 3) was coupled to both cloxicillin and carbenicillin sodium to give N-methylated hydroxamates 15 and 16.
All cephalosporins were tested for antibacterial activity using the agar diffusion assay, with MICs determined for select compounds.11, 12 As shown in Table 1, many compounds exhibited notable activity against Gram-positive bacteria, albeit less than their parent counterparts (that is, featuring free carboxylate). Highlighted among the compounds was the activity of cephalosporin 2b, with MIC values of 0.2 μM, 3 μM, and 3 μM against B. subtilis, S. aureus and M. luteus, respectively. Further, compound 3, featuring a free hydroxamic acid exhibited good activity, with MIC values of 0.4 μM and 1.6 μM against B. subtilis and S. aureus, respectively. Although most of the compounds were not very active against Gram-negative bacteria, cephalosporin 4a, featuring the ATMO sidechain, showed good activity against Pseudomonas aeruginosa (K799/61) and both strains of Escherichia coli tested. When analyzing how N-substitution affected activity, cephalosporins bearing N-methyl substituted hydroxamates exhibited, in general, a decrease in both potency and/or broadness of antibacterial activity. This is illustrated by comparing the activities of 4b with 9 and 2a with 10.
Similarly, penicillins were tested via diffusion assays with MIC values determined for select compounds (Table 2). Hydroxamate containing penicillins were quite active against Gram-positive microbes as well, although generally not as active as the parent compounds. Notably, compounds 11 and 13 demonstrated significant activity against S. aureus, with MIC values of 0.4 μM and 1.6 μM, respectively. Interestingly, the rearranged urea, 12, had good activity against Gram-positive rods and activity against E. coli DC2 with a zone of inhibition of 26 mm. Removal of the acidic proton by N-methylation only slightly affected the activity of cloxicillin derivatives (compounds 13 and 15) but had a substantial effect on the carbenicillins, as replacing both carboxylates with hydroxamates gave a compound (that is, N-methyl hydroxamate 16) that was completely inactive.
As shown in Table 3, select compounds were also screened against three S. aureus strains (SG511, ISR-001 and ISR-002). All compounds tested showed notable zones of inhibition, ranging from 22 to 32 mm. With these encouraging results, MIC values were also determined. Of special interest were the values for compounds 11 and 13, with MICs as low as 0.4 μM for both compounds. Lastly, carbenicillin analogue 14 exhibited MICs in the range of 1.6–6 μM against all three strains, while the parent compound was seen to be inactive against strains ISR-001 and ISR-002.
To close, we have synthesized a focused set of cephalosporins and penicillins with hydroxamate replacement of the classical carboxyl groups. Preliminary biological assessments indicated that several of the hydroxamates retained broad spectrum activity against Gram-positive (including select MRSA strains) and Gram-negative pathogens. Further, removal of the acidic proton of the hydroxamate portion diminished both level of and the spectrum of activity. Thus, further functionalization of the ionizable position of these important antibiotics might be of value in development of much needed alternative forms of antibiotics.

Syntheses of hydroxamate cephalosporins 2–3.

Syntheses of cephalosporins with ATMO side-chain.

Syntheses of N-methyl O-alkyl hydroxylamines 7a–b.

Syntheses of N-methyl hydroxamate cephalosporins 8–10.

Initial coupling attempt with cloxicillin.

Improved coupling of penams under acidic aqueous conditions.

Coupling of 7a to penams.
References
Page, M. I. & Jaws, A. P. The chemical reactivity of β-lactams, β-sultams and β-phospholactams. Tetrahedron 56, 5631–5638 (2000).
Varetto, L., Frere, J. M. & Ghuysen, J. M. The importance of the negative charge of β-lactam compounds for the inactivation of the active-site serine DD-peptidase of Streptomyces R61. FEBS 225, 218–222 (1987).
Miller, M. J., DeBons, F. E. & Loudon, G. M. The chemistry of a method for the determination of carboxyl-terminal residues in peptides. J. Org. Chem. 42, 1750–1762 (1977).
Brown, L. D., Zygmunt, W. A. & Stavely, H. E. Active derivatives of penicillin. App.Microbiol 17, 339–343 (1969).
Godfrey, J. C. (Bristol-Myers Co.) N-Hydroxypenicillinamides. US Patent 330181119670131.
Lewis, B. A., Sassiver, L. M. & Shepherd, R. G. (American Cyanamid Company) 7-(Phenylacetylamino) Cephalosporin Carboxamides and 7-(Thiophene-2-Acetylamino) Cephalosporin Carboxamides. US Patent 3641015A19720208.
Desikan, P. & Amberg, C. H. Catalytic hydrodesulfurization of thiophene. V. hydrothiophenes. selective poisoning and acidity of the catalyst surface. Can. J. Chem. 42, 843–850 (1964).
Woulfe, S. R. & Miller, M. J. Synthesis and biological activity of substituted [[3(S-(acylamino)-2-oxo-1-azetidinyl]oxy] acetic acids. a new class of heteroatom-activated β-lactam antibiotics. J. Med. Chem. 28, 1447–1453 (1985).
Ramasamy, K., Olsen, R. K. & Emery, T. N-Methylation of O-benzyl-α-N-(alkoxycarbonyl)-α-amino acid hydroxamate derivatives. J. Org. Chem 46, 5438–5441 (1981).
Sharma, S. K., Miller, M. J. & Payne, S. M. Spermexatin and spermexatol: new synthetic spermidine-based siderophore analogues. J. Med. Chem 32, 357–367 (1989).
Afonin, S. et al. 4-fluorophenylglycine as a label for 19F NMR structure analysis of membrane-associated peptides. Chem. Bio.Chem 4, 1151–1163 (2003).
Murray, P. R., Baron, E. J., Pfaller, M. A., Tenover, F. C. & Yolken, R. H. Manual of Clinical Microbiology, American Society for Microbiology, Washington, DC, USA, (1999).
Acknowledgements
We acknowledge the University of Notre Dame, and partial funding from the NIH (NIH-2R01-AI054193-05A2) for support of this work. We acknowledge Nonka Sevova (Mass Spectrometry and Proteomics Facility, UND) for mass spectroscopic analyses and Viktor Krchnak (UND) for LC/MS analysis. MWM acknowledges an ECK Global Health Fellowship (2014–2015, UND).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Supplementary information
Rights and permissions
About this article

Cite this article
Majewski, M., Miller, P. & Miller, M. Studies at the ionizable position of cephalosporins and penicillins: hydroxamates as substitutes for the traditional carboxylate group. J Antibiot 70, 292–296 (2017). https://doi.org/10.1038/ja.2016.149
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2016.149
This article is cited by
-
Photon and vibration synergism on planar defects induced 2D-graphitic carbon nitride for ultrafast remediation of dyes and antibiotic ampicillin
Journal of Materials Science (2022)
