
Abstract
Recent studies have expanded the phylum Chlorobi, demonstrating that the green sulfur bacteria (GSB), the original cultured representatives of the phylum, are a part of a broader lineage whose members have more diverse metabolic capabilities that overlap with members of the phylum Bacteroidetes. The 16S rRNA gene of an uncultivated clone, OPB56, distantly related to the phyla Chlorobi and Bacteroidetes, was recovered from Obsidian Pool in Yellowstone National Park; however, the detailed phylogeny and function of OPB56 and related clones have remained unknown. Culturing of thermophilic bacterial consortia from compost by adaptation to grow on ionic-liquid pretreated switchgrass provided a consortium in which one of the most abundant members, NICIL-2, clustered with OPB56-related clones. Phylogenetic analysis using the full-length 16S rRNA gene from NICIL-2 demonstrated that it was part of a monophyletic clade, referred to as OPB56, distinct from the Bacteroidetes and Chlorobi. A near complete draft genome (>95% complete) was recovered from metagenomic data from the culture adapted to grow on ionic-liquid pretreated switchgrass using an automated binning algorithm, and this genome was used for marker gene-based phylogenetic analysis and metabolic reconstruction. Six additional genomes related to NICIL-2 were reconstructed from metagenomic data sets obtained from thermal springs at Yellowstone National Park and Nevada Great Boiling Spring. In contrast to the 16S rRNA gene phylogenetic analysis, protein phylogenetic analysis was most consistent with the clustering of the Chlorobea, Ignavibacteria and OPB56 into a single phylum level clade. Metabolic reconstruction of NICIL-2 demonstrated a close linkage with the class Ignavibacteria and the family Rhodothermaceae, a deeply branching Bacteroidetes lineage. The combined phylogenetic and functional analysis of the NICIL-2 genome has refined the membership in the phylum Chlorobi and emphasized the close evolutionary and metabolic relationship between the phyla Chlorobi and the Bacteroidetes.
Similar content being viewed by others


Introduction
Access to uncultivated populations through metagenomics and single cell sequencing has greatly expanded our knowledge of microbial diversity (Tyson et al., 2004; Dick et al., 2009; Hess et al., 2011; Mackelprang et al., 2011; Dupont et al., 2012; Wrighton et al., 2012; Albertsen et al., 2013; Rinke et al., 2013; Sharon et al., 2013). Genomic sequences of the uncultivated microbial populations recovered with these methods have significantly expanded the tree of life, in which half of the phyla or divisions do not have cultivated representatives (Rappe and Giovannoni, 2003; Rinke et al., 2013). Defining the phylogeny and function of these uncultivated organisms is critical to understand the roles these microbes have in their native ecosystems and how they may be employed in biotechnology.
The phylum Chlorobi was originally classified based on the properties of the green sulfur bacteria (GSB), which are defined as the family Chlorobiaceae within the class Chlorobea (Imhoff, 2003). The GSB are strictly anaerobic, non-motile, obligate phototrophs that oxidize reduced sulfur compounds for CO2 fixation via the reverse tricarboxylic acid (rTCA) cycle and can perform N2 fixation (Buchanan and Arnon, 1990; Wahlund and Madigan, 1993; Wahlund and Tabita, 1997). The sole exception is Chlorobium ferrooxidans, which instead uses ferrous iron as a reductant and can conduct assimilatory sulfate reduction (Heising et al., 1999; Overmann, 2008; Frigaard and Dahl, 2009; Gregersen et al., 2011). Close phylogenetic relationships based on 16S rRNA gene surveys between the Chlorobi and the Bacteroidetes/Fibrobacter phyla, characterized as chemoheterotrophs with broad geographic distributions, led to the establishment of the FCB (Fibrobacter-Chlorobi-Bacteroidetes) superphylum (Woese et al., 1985; Gupta, 2004). Phylogenetic analysis based on genomes obtained by reconstruction from metagenomic data sets and single cell sequencing has further expanded the FCB superphylum to include poorly characterized (Gemmatimonadetes, Caldithrix) and candidate phyla (Zixibacteria, Marinomicrobia, WS3, WWE1) (Castelle et al., 2013; Rinke et al., 2013).
Recently, the phylum Chlorobi was expanded by the reconstruction of the genome of ‘Candidatus Thermochlorobacter aerophilum’, which was recovered from a metagenomic data set obtained from Yellowstone National Park hot springs (Liu et al., 2012b). Unlike the GSB, evidence was obtained for a photoheterotrophic lifestyle for ‘Ca. T. aerophilum’, and no genes were detected in the reconstructed genome for sulfur oxidation, N2 fixation or the rTCA cycle. Genomic analysis of two chemoheterotrophic thermophilic isolates, Ignavibacterium album and Melioribacter roseus, indicated that they form a deeply branching lineage, the class Ignavibacteria, near the base of the Chlorobi (Liu et al., 2012a; Kadnikov et al., 2013; Podosokorskaya et al., 2013). The Ignavibacteria genomes do not encode the photosynthetic apparatus typical of the Chlorobi, but retain most of the genes encoding for the rTCA cycle. Based on phylogeny and function, the Ignavibacteria has been proposed as either a novel class in the Chlorobi (Liu et al., 2012a) or a distinct phylum (Podosokorskaya et al., 2013).
Surveys based on 16S rRNA genes have found an uncultivated lineage affiliated with the Chlorobi that is more deeply branching than the Ignavibacteria. The earliest identified sequence in this lineage belonged to OPB56, a clone that was first recovered from the Obsidian Pool in Yellowstone National Park (Hugenholtz et al., 1998). Related clones also have been recovered from high temperature environments such as hot springs and compost piles (Fouke et al., 2003; Ross et al., 2012). Culturing thermophilic bacterial consortia to grow with biomass substrates as the sole carbon source was initiated to discover new glycoside hydrolases (Gladden et al., 2011; Park et al., 2012; D'haeseleer et al., 2013) and understand community dynamics during biomass deconstruction (Eichorst et al., 2013, 2014). Here, we report the recovery of a draft genome of a representative of the OPB56 lineage from metagenomic data obtained from a thermophilic microbial consortium adapted to grow on switchgrass pretreated with 1-ethyl-3-methyimidazolium acetate, a promising ionic liquid for biomass pretreatment. Phylogenetic and functional analyses of the genome of this uncultivated population have refined our understanding of the phylum Chlorobi and its relationship with the Bacteroidetes.
Materials and methods
Sample collection and microbial community enrichment
Sample collection and enrichment procedures were described previously (Eichorst et al., 2013, 2014). Briefly, compost samples were collected from a municipal green waste composting facility, the Newby Island Sanitary Landfill in Milpitas, California. Replicate thermophilic microbial communities were enriched on switchgrass pretreated with ionic liquid 1-ethyl-3-methylimidazolium acetate (Dibble et al., 2011) as the sole carbon source at 60 °C (Eichorst et al., 2014). The enrichment was serially transferred through four passages and DNA was extracted as previously described (D'haeseleer et al., 2013).
Sequencing and assembly of metagenomic reads
DNA fragments for Illumina sequencing were created using the Joint Genome Institute standard library generation protocols for Illumina HiSeq 2000 platforms (Wu et al., 2014). Illumina sequencing was performed on a HiSeq 2000 system. Raw reads were trimmed using a minimum quality cutoff of Q10. Trimmed, paired-end Illumina reads were assembled using SOAPdenovo v1.05 (http://soap.genomics.org.cn/soapdenovo.html) with default settings (−d 1 and −R) at different Kmer sizes (85, 89, 93, 97, 101 and 105, respectively). Contigs generated by each assembly (a total of six contig sets from the six k-mer sizes) were merged using in-house Perl scripts as following. Contigs were first dereplicated and sorted into two pools based on length. Contigs <1800 bp were assembled using Newbler (Life Technologies, Carlsbad, CA, USA) to generate larger contigs (−tr, −rip, −mi 98, −ml 80). All assembled contigs >1800 bp, as well as the contigs generated from the Newbler assembly, were combined and merged using minimus2 (−D MINID=98 −D OVERLAP=80) (AMOS: http://sourceforge.net/projects/amos). The average fold coverage (or read depth) of each contig was estimated by mapping all Illumina reads back to the final assembly using BWA (version 1.2.2) (Li and Durbin, 2009).
Binning of assembled metagenomic scaffolds
The assembled contigs were binned using MaxBin (Wu et al., 2014) (http://downloads.jbei.org/data/MaxBin.html) with default setting (minimum contig length=1000 bps). The tool considers both tetramer frequency profiles and abundance levels of the contigs and uses an expectation-maximization algorithm to classify the contigs. The phylogeny of the binned genomes was assigned using NCBI BLAST (Altschul et al., 1997) and MEGAN (Huson et al., 2011).
Identifying OPB56 lineages in thermal spring metagenomic data sets
Two fosmid clones obtained from a 70 °C subterranean thermal spring in a Japanese gold mine, JFF029_C06 (NCBI acc. AP011722) and JFF027_B02 (NCBI acc. AP011715), had 22 ribosomal protein sequences >80% identical to the sequences from NICIL-2 (Nunoura et al., 2005). Ten metagenomic data sets sampled from thermal environments were chosen from the IMG/M website (https://img.jgi.doe.gov/cgi-bin/m/main.cgi) using ‘Yellowstone’ or ‘hot spring’ as keywords. The assembled sequences from these data sets were binned using MaxBin with default options, and 22 ribosomal proteins shared among the NICIL-2 genome and the 2 thermal spring fosmid clones (JFF029_C06 and JFF027_B02) were used as markers to identify genome bins that were closely related to the OPB56 lineage (>80% amino-acid identity). In total, six genome bins that possess all 22 ribosomal proteins were identified from four metagenomic data sets. Two genomes were obtained from a sample from the upper layer of an anoxygenic and chlorotrophic microbial mat collected at Mushroom Spring, Yellowstone National Park (JGI IMG/M taxon ID 3300002510). Two genomes were recovered from a sample obtained from Obsidian Pool at Yellowstone (JGI IMG/M taxon ID 3300002966). One genome was recovered from a sample from a hypersaline microbial mat at Yellowstone Fairy Falls (JGI IMG/M taxon ID 3300003606). One genome was recovered from a sample from a cellulolytic enrichment at 77 °C with water from Nevada Great Boiling Spring (JGI taxon IMG/M ID 3300000083).
Phylogenetic trees for NICIL-2
Phylogenetic trees were built using 16S ribosomal RNA genes. Homologs of the NICIL-2 16S rRNA gene were identified by BLASTN in the Geneious 6.1 software (Biomatters, http://www.geneious.com) and 16S rRNA genes of Bacteroidetes, and Chlorobi were collected from NCBI websites (http://www.ncbi.nlm.nih.gov/). 16S rRNA sequences of two homologs were recovered from whole fosmid sequences JFF029_C06 (NCBI acc. AP011722) and JFF027_B02 (NCBI acc. AP011715). The 16S rRNA sequence Fibrobacter succinogenes S85 was used as the outgroup. In total, 65 sequences were used to build the 16S gene tree. A total of 14 nucleotide 16S rRNA phylogenetic trees were constructed with a MUSCLE alignment (Edgar, 2004) trimmed in GBlocks (Talavera and Castresana, 2007), three statistical methods (Maximum Likelihood, Neighbor-Joining and Minimum Evolution) and several substitution methods (General Time Reversible, Kimura 2-parameter, Tamura-Nei, Tamura 3-parameter, Jukes-Cantor, Number of differences and p-distance) in MEGA5 (Tamura et al., 2011). Where applicable, non-uniformity of evolutionary rates among sites was modeled by using a discrete Gamma distribution (0.3) with five rate categories and by assuming that a certain fraction of sites are evolutionarily invariable. Reliability of inferred trees was conducted with 1000 bootstrap replicates. A maximum likelihood consensus tree built upon the General Time Reversible was selected as the consensus tree. An expanded phylogenetic tree depicting all nodes can be found in Supplementary Figure S1. Nodes without bootstrap values have <50% consensus. Temperatures of sampling environments used to color code the 16S tree were complied from metadata submitted to GenBank along with clone sequences. Where applicable, sampling temperatures were verified by publication. Clones derived from high (55–80 °C) or moderate (20–32 °C) temperature environments were colored red or green, respectively. Clones lacking metadata regarding temperature were left uncolored (black).
Next, a phylogenetic tree was built based on 22 ribosomal proteins that were shared among the isolate genomes from the Bacteriodetes and Chlorobi, the six genome bins recovered from four metagenomes from thermal springs, and two thermal spring fosmid clones (Supplementary Table S1). The protein sequences of the 22 ribosomal proteins were aligned separately using MUSCLE (Edgar, 2004), concatenated using customized PERL script, and refined using Gblocks (Talavera and Castresana, 2007). The tree was built in MEGA5 (Tamura et al., 2011) using maximum likelihood estimation with bootstrap value set to 1000. Settings for building concatenated protein tree in MEGA5 were JTT model, uniform evolutionary rates among all sites and complete deletion. All other options were set to default values.
A tree based on 86 single copy proteins was also constructed to further validate the tree built from the 22 ribosomal proteins (proteins listed in Supplementary Table S2). The protein-coding genes of NICIL-2 and the isolate Bacteroidetes and Chlorobi genomes that were involved in constructing the 22-ribosomal-protein tree were searched against the Hidden Markov models of all protein families, which were downloaded from the PFAM website (Finn et al., 2014), using HMMER3 (http://hmmer.janelia.org/) (Mistry et al., 2013) with E-value cutoff set to 1e−5. A phylogenetic tree was built using 86 protein families that appeared only once in every species with exactly the same procedures and settings as the 22-ribosomal-protein tree.
Finally, components of the electron transport chain were used to infer relationships between clades. Homologs of Complex I and alternative Complex III subunits were used to build unrooted neighbor joining trees. Subunits were concatenated and aligned with MUSCLE using Geneious software. Trees were built in MEGA 5.0 based on the JTT substitution model with 1000 bootstrap replicates.
Metabolic analysis of NICIL-2
The extracted NICIL-2 genome was uploaded to RAST server (http://rast.nmpdr.org/) (Overbeek et al., 2014) for annotation. Pathway analysis of NICIL-2 was performed by using KEGG2 KAAS genome annotation web server (Moriya et al., 2007) and visualized by KEGG2 Search-and-Color-Pathway Mapper (http://www.genome.jp/kegg/tool/map_pathway2.html). Pathway holes were filled by using NCBI BLAST to search for proteins that were missing in the visualized KEGG2 Pathway results or RAST annotations.
Extraction of carbohydrate active enzymes
Carbohydrate active enzymes, described by the CAZy database (Lombard et al., 2014), were extracted using dbCAN (Yin et al., 2012). Cellulases (GH5, 6, 7, 9, 44, 45, 48), hemicellulases (GH8, 10, 11, 12, 26, 28, 53) and oligosaccharide-degradation enzymes (GH1, 2, 3, 29, 35, 38, 39, 42, 43, 52) were classified following previously described criteria (Allgaier et al., 2010).
Data availability
Metagenomic data for the thermophilic bacterial consortium adapted to grow on ionic-liquid treated switchgrass is available on the IMG website (https://img.jgi.doe.gov/cgi-bin/m/main.cgi) (Markowitz et al., 2014) with taxon object ID 3300000145. The sequences for the NICIL-2 genome have been deposited at DDBJ/EMBL/GenBank under the accession LDXS00000000. The version described in this paper is version LDXS01000000. The NICIL-2 RAST annotation, the six OPB56 genome bins extracted from the thermal metagenomes and their RAST annotations are available at http://downloads.jbei.org/data/microbial_communities/microbial_communities.html.
Results
Genomic reconstruction of NICIL-2
Microbial community compositional analysis of thermophilic bacterial consortia adapted to grow on biomass substrates (cellulose, xylan and switchgrass) as their sole carbon source at 60 °C consistently identified a ubiquitous OTU97 that was distantly related to cultured members of the phylum Chlorobi (Eichorst et al., 2013, 2014). Metagenomic sequencing of a consortium adapted to grow on ionic-liquid pretreated switchgrass, in which the Chlorobi-related OTU97 was abundant, was performed to understand the phylogeny and metabolic potential of the population that was represented by this OTU97. Assembly (see Materials and methods) and automated binning (Wu et al., 2014) of the metagenome of this consortium yielded 20 genomic bins (Supplementary Table S3), among which the most abundant bins were a population closely related to Chitinophagaceae strain NYFB (61.8%), which was isolated from a related enrichment grown on cellulose (Eichorst et al., 2013), and the Chlorobi-related bin (23.1%). Bins present at >1% included multiple uncultured populations clustering with the Paenibacillaceae (bins 003 and 006), an uncultivated population clustering with Verrucomicrobia subdivision 3 (bin 004) and a population closely related to the Thermobipora bispora (bin 005), an actinobacterial thermophile.
The Chlorobi-related bin was named NICIL-2 (for Newby Island Compost Ionic Liquid-2nd in abundance). Analysis of the proteins predicted from NICIL-2 genome was consistent with its identification as a population distantly related to members of the FCB superphylum, with the Bacteroidetes (33%) and Ignavibacteria (15.7%) having the most closely related protein sequences (Figure 1). The recovered NICIL-2 draft genome was relatively small (2.67 Mbps) and near complete (95.3%; 102 out of 107 single copy marker genes in 152 scaffolds). The N50 length for the NICIL-2 draft genome was 168 929 bp and the largest contig was 1.1 MB, suggesting that most of the scaffolds represented a high-quality assembly (Table 1).

The distribution of the best-matched lineage for all NICIL-2 proteins. Percentages are estimated by dividing protein counts that matched to each lineage against the number of all NICIL-2 proteins. See Materials and methods for details.
Phylogenetic analysis of NICIL-2
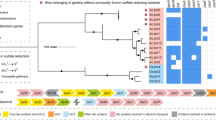
A 16S rRNA gene (1451 bp) was recovered from the NICIL-2 draft genome. The ribotype most closely related to the 16S rRNA gene of NICIL-2 (>99% identical) was sequenced from a fosmid clone (JFF029_06) recovered from a thermal stream (70 °C) in a Japanese gold mine sequence (Nunoura et al., 2005). 16S rRNA genes from cultured representatives of the Bacteroidetes and Chlorobi were <85% identical to the NICIL-2 sequence. A phylogenetic tree constructed by aligning 16S rRNA gene sequences related to NICIL-2 demonstrated that the lineage containing NICIL-2 was distinct from the Bacteroidetes and Chlorobi (Figure 2). The NICIL-2 lineage contained two lines of descent based on temperature of the sampling environment. The high temperature cluster (depicted in red in Figure 2) included ribotypes primarily recovered from high temperature environments ranging from 55 °C to 80 °C, such as the clone OPB56 (Hugenholtz et al., 1998). The second cluster included ribotypes primarily recovered from moderate temperature environments ranging from 20 °C to 32 °C (depicted in green in Figure 2). Since OPB56 was the first clone discovered that is affiliated with this phylogenetic cluster, the lineage containing NICIL-2 is referred to as the OPB56 clade. An expanded view of the 16S rRNA gene tree is depicted in Supplementary Figure S1.

The maximum likelihood phylogenetic tree built for the novel lineage NICIL-2 using the 16S ribosomal RNA gene. The clade closest to species NICIL-2 was from a fosmid isolated from a Japanese gold mine (Nunoura et al., 2005). Species temperature ranges were determined from the literature associated with each NCBI accession number. The inhabitant temperature for species was marked by red (>55 °C), green (20–32 °C) or black (undetermined). Scale bar denotes 0.05 changes per nucleotide site. Details for tree building are provided in Materials and methods. The expanded phylogenetic tree showing all nodes can be found in Supplementary Figure S1. A full colour version of this figure is available at the ISME Journal online.
To establish the phylogenetic affiliation of NICIL-2 and the OPB56 clade more completely, additional ribosomal protein sequences were obtained from data recovered from natural samples. Homologs of 22 conserved ribosomal genes in NICIL-2 were found in two Japanese thermal spring fosmid clones (JFF029_C06 and JFF027_B02). This set of 22 ribosomal proteins was used to search metagenomic data sets from high temperature environments. Complete sets of these conserved ribosomal genes were identified in four metagenomic data sets obtained from high temperature environments. Automated binning of these data sets recovered six near-complete (>90% complete) draft genomes that contained sequences for the conserved 22 ribosomal proteins in the NICIL-2 genome and the Japanese gold mine fosmid clones (Supplementary Table S4). Five of the six concatenated protein sequences clustered with in the OPB56 lineage with NICIL-2, while one of sequences from Yellowstone Fairy Falls clustered with the Ignavibacteria (Figure 3a). This phylogenetic tree demonstrated that the Chlorobea, Ignavibacteria and OPB56 formed a monophyletic clade with high confidence (97%). The Bacteroidetes formed a distinct cluster, and the family Rhodothermaceae, whose affiliation with the Bacteroidetes has been questioned (Nolan et al., 2009), were affiliated with the Bacteroidetes with high confidence (>80%). A second phylogenetic tree was constructed by concatenating an alignment of 86 single copy genes shared among Bacteroidetes, Chlorobi and Fibrobacter (Figure 3b). This tree reproduced the topology observed for the gene built from the 22 ribosomal genes, further supporting the affiliation of the Chlorobea, Ignavibacteria and OPB56.

The maximum likelihood phylogenetic tree built for the novel lineage NICIL-2 using (a) 22 ribosomal proteins and (b) 86 single-copy proteins shared among Bacteroidetes, Chlorobea, Ignavibacteria, OPB56 and Fibrobacter clusters. Scale bar denotes 0.1 changes per amino-acid site. Details for tree building are provided in Materials and methods.
A complementary approach to understanding evolutionary relationships between Bacteriodetes and Chlorobi has been to identify indels in conserved proteins (Gupta and Lorenzini, 2007). Insertions in DNA polymerase III (28 aa) and alanyl-tRNA synthetase (12–14 aa) that are conserved among the GSB and absent among the Bacteriodetes were attributed as characteristic of the phylum Chlorobi. Alignments of these two proteins (Supplementary Figures S2 and S3) indicate that the DNA polymerase III insertion in the GSB protein sequences is not present in the predicted proteins from the Ignavibacteriae and NICIL-2 genomes, while 2–3 amino acids of the insertion into the GSB alanyl-tRNA synthetase sequences are conserved in the Ignavibacteria and NICIL-2 protein sequences.
Metabolic reconstruction of NICIL-2
The physiology of NICIL-2 was inferred by metabolic reconstruction and its metabolic potential compared to representatives of GSB (Chlorobaculum tepidum), Bacteroidetes (Rhodothermus marinus and Salinbacter ruber) and Ignavibacteria (Ignavibacterium album and Meliobacter roseus). Genes encoding photosynthesis-related proteins, including homologs of C. tepidum’s photosynthetic reaction center subunits (CT1020, pscB, psC, pscD), chlorosome envelope proteins (csmABCDEFHIJX), and bacteriochlorophyll a proteins (fmoA), are absent in all the other genomes, distinguishing the GSB in this comparative set (Eisen et al., 2002). A visual summary of the metabolic reconstruction is presented in Figure 4. For full gene information, box numbers and abbreviations, see Supplementary Tables S5 and S6.

Reconstructed metabolism of NICIL-2 inferred from the reassembled genome. For full gene information, box numbers and abbreviations, see Supplementary Tables S5 and S6. Red text represents enzymes or biosynthetic pathways that are missing from the genome. Blue, green and purple arrows indicate ATP, NADH and NADPH flow, respectively.
Carbon metabolism
The NICIL-2 genome encodes a complete set of genes for glycolysis, the TCA cycle and gluconeogenesis (Figure 4). Genes for the rTCA cycle, which are present and expressed for autotrophic carbon fixation in the GSB, are absent. Furthermore, the presence of genes encoding lipoic acid-containing cofactors in the pyruvate dehydrogenase and α-ketoglutarate dehydrogenase complexes suggests that the TCA cycle functions in the oxidative direction. Most of the rTCA cycle has been reconstructed in R. marinus and I. album, including multiple pyruvate-ferredoxin oxidoreductases and α-ketoglutarate-ferredoxin oxidoreductases, two required enzymes for the rTCA cycle. The I. album and R. marinus genomes lack ATP-dependent citrate lyase, a critical enzyme to complete the rTCA cycle (Buchanan and Arnon, 1990). It has been proposed that I. album may use an ATP-independent citrate lyase to function in the rTCA cycle instead, although this claim is untested experimentally and neither I. album nor R. marinus have been shown to grow autotrophically (Liu et al., 2012a).
Despite its high relative abundance in adapted cultures growing on plant biomass, NICIL-2 had a surprisingly limited enzymatic capacity to deconstruct complex biomass. The comparison of the metabolic potential for polysaccharide hydrolysis among the 20 bins extracted from the metagenome from pretreated-switchgrass enrichment demonstrated that NICIL-2 has relatively fewer genes for cellulose and hemicellulose deconstruction compared with other members of the microbial community (Supplementary Figure S4). In particular, strain NYFB, the verrucomicrobial population and multiple Gram-positive Firmicutes have more extensive repertories of genes for polysaccharide hydrolysis. Additional inspection of the reconstructed OPB56-affiliated genomes from high temperature natural samples demonstrated that the lack of genes for polysaccharide hydrolysis was common to this clade. Among the genomes clustering with the Bacteroidetes and Chlorobi, R. marinus, M. roseus and Yellowstone Obsidian Pool Bin 062, which clusters with the Ignavibacteria, possessed an extensive repertoire of glycoside hydrolases to deconstruct plant biomass. These analyses suggest that NICIL-2 and related members of the OPB56 clade are probably not involved in the primary deconstruction of biomass in the adapted community and natural environments. It is conceivable that NICIL-2 may grow on sugar monomers or oligomers; however, only one predicted disaccharide transporter gene was identified in the genome.
Although the selection for NICIL-2 occurred under aerobic conditions, it may possess the capacity for fermentation. Genes for the fermentative production of ethanol are present (via alcohol dehydrogenase, EC 1.1.1.1), while genes for formate (via pyruvate formate lyase, EC 2.3.1.54), lactate (via lactate dehydrogenase, EC 1.1.1.27) and propionate (via methylmalonyl-CoA carboxyl transferase, 2.1.3.1) are absent. The NICIL-2 genome encodes a gene for phosphotransacetylase (EC 2.3.1.8), but not acetate kinase (EC 2.7.2.1), both of which are required for fermentative acetate production. Both I. album and M. roseus contain genes for the production of lactate and acetate, while C. tepidum does not.
Nitrogen and sulfur metabolism
Genes for ammonium assimilation, glutamine synthase (EC 6.3.1.2) and glutamate synthase (EC 1.4.1.13) were detected in the NICIL-2 genome. However, no genes were identified for dissimilatory nitrate reduction, assimilatory nitrate reduction, denitrification, nitrogen fixation and nitrification. C. tepidum encodes nif genes required for N2 fixation, while M. roseus (Kadnikov et al., 2013), I. album (Liu et al., 2012a) and R. marinus (Nolan et al., 2009) do not. C. tepidum is an obligate sulfur oxidizer and therefore can oxidize sulfide, thiosulfate, sulfite and elemental sulfur. C. tepidum preferentially oxidizes sulfide to elemental sulfur, then elemental sulfur and thiosulfate to sulfate (Chan et al., 2008). Additionally, sulfite may be oxidized when supplied in the growth medium, but it cannot sustain C. tepidum as the sole electron donor (Rodriguez et al., 2011). NICIL-2, I. album, M. roseus and R. marinus lack all genes required for the oxidation of reduced sulfur compounds.
Amino-acid biosynthesis
The NICIL-2 genome is missing many key genes for the biosynthesis of amino acids. Complete pathways are encoded for alanine, arginine, asparagine, aspartate, β-alanine, glutamate, glycine, lysine and methionine. NICIL-2 lacks ilvC and leuABCD and therefore possesses incomplete pathways for valine, leucine and isoleucine biosynthesis. Similarly, the I. album genome lacked all genes required for valine, leucine and isoleucine biosynthesis from pyruvate except for the branched-chain amino transferase (ilvE), while M. roseus and C. tepidum contain complete pathways. Neither NICIL-2 nor I. album encodes genes required for proline biosynthesis from glutamate (proBAC), while both C. tepidum and M. roseus do. The serB gene for biosynthesis of serine is missing in NICIL-2, I. album, M. roseus, and is found in some GSB, including C. tepidum. NICIL-2 may also use amino acids as growth substrates. Complete degradation pathways are present for branched-chain amino acids (leucine, isoleucine and valine), similar to pathways observed in ‘Candidatus Thermochlorobacter aerophilum’ (Liu et al., 2012b).
Electron transport
The major electron transport chain components were identified in the NICIL-2 genomes including: NADH:ubiquinone oxidoreductase (Complex I, EC 1.6.5.3), membrane-bound succinate dehydrogenase (Complex II, EC 1.3.5.1), quinol-oxidizing alternative Complex III (ACIII) (Yanyushin et al., 2005; Pereira et al., 2007), several cytochrome c oxidases (Complex IV, EC 1.9.3.1) and an F-type H+-transporting ATPase (Complex V, EC 3.6.3.14). NICIL-2 contains one complete set of genes for NADH:ubiquinone oxidoreductase (14 subunits, nuoABCDEFGHIJKLMN), although they are not assembled in one operon (Supplementary Figure S6). Instead, the majority of genes are arranged independently throughout the largest NICIL-2 scaffold (nuoGHI, nuoJK, nuoF, nuoD, nuoE and nuoMN), and the remaining genes are found on three smaller scaffolds (nuoAB, nuoL and nuoC), indicating that the absence of an operon structure is not an artifact of assembly. C. tepidum contains one set of genes encoding for the NADH:ubiquinone oxidoreductase that lacks nuoEFG (11 subunits). I. album and M. roseus contain two sets of nuoABCDHIJKLMN and one set of nuoEFG each (Supplementary Figure S6). Interestingly, Complex I from NICIL-2 is most closely related to those of Rhodothermus marinus and Salinibacter ruber of the Bacteroidetes, both of which contain one complete set of genes for NADH:ubiquinone oxidoreductase (Figure 5a). Homologs for the 11 Complex I subunits were also identified in all six NICIL-2 related bins recovered from Yellowstone and Great Boiling Spring, and a concatenated assembly of these protein sequences clustered with the sequences from NICIL-2.

Neighbor joining unrooted concatenated protein trees of Complex I (a) and alternative Complex III (b). Numbers in parentheses denote the number of subunits used to build the tree. Scale bar denotes x changes per amino-acid site.
The ACIII is a new class of bacterial membrane oxidoreductases found in organisms that often lack the bc1 complex (Yanyushin et al., 2005). The ACIII from R. marinus (Pereira et al., 2007; Refojo et al., 2013) and filamentous anoxygenic phototrophic bacterium Chloroflexus aurantiacus (Gao et al., 2009, 2013) has been purified and studied. Recently, large numbers of gene clusters coding for ACIII subunits, with variations in constitution and organization, were found in a range of bacterial genomes (Refojo et al., 2013). For example, R. marinus contains the genes actABCDEF in an operon that encodes six ACIII subunits; homologs of this gene cluster have been identified in multiple members of the Bacteroidetes (Thiel et al., 2014). A phylogenetic tree depicts the relationship of ACIII from NICIL-2 to its closest relatives (Figure 5b). The GSB do not possess ACIII and therefore they are not represented in this tree. However, ‘Candidatus Thermochlorobacter aerophilum, which is not a member of the GSB, encodes an ACIII that likely functions in aerobic respiration (Liu et al., 2012b). It is important to note that both I. album and M. roseus are unique among this set of ACIII as they contain five subunits, with the actDE subunits identified as a fusion protein. A full complement of genes coding for all the ACIII subunits, which were recovered from two NICIL-2-related genome bins from the Yellowstone metagenomes, were found to contain the actDE fusion and clustered with I. album and M. roseus. NICIL-2 does not contain this fusion and the genes for its ACIII Complex are more closely related to R. marinus and S. ruber.
NICIL-2 terminal electron acceptors include a type-aa3 cytochrome c oxidase (Complex IV) and possible alternative cytochrome c oxidases, annotated as CoxMOP. NICIL-2 is unlikely to be microaerophilic because type cbb3 oxidases were not detected in the genome. Additionally, cytochrome bd was undetected. For comparison, both I. album and M. roseus genomes contain genes for cbb3 type cytochrome c oxidase and a cytochrome bd complex.
The NICIL-2 genome contains an incomplete set of men genes for the synthesis of menaquinone. A complete menaquinone biosynthetic pathway contains menFDHCEBAG (Bentley and Meganathan, 1982) and the NICIL-2 genome only includes menBAG. For comparison, C. tepidum and I. album contain complete men pathways, while the M. roseus and R. marinus genomes have annotations for all required genes except menB and menH, respectively. Homologs of genes encoding enzymes of the alternative menaquinone biosynthetic pathway, the futalosine pathway (Arakawa et al., 2011), were undetected in NICIL-2. Ubiquinone biosynthetic genes are also absent. The NICIL-2 genome may have incomplete coverage in this area and additional men genes may be found with a completely assembled genome. Alternatively, NICIL-2 may engage in quinone exchange with other community members, as observed for the phototrophic consortium ‘Chlorochromatium aggregatum’ (Liu et al., 2013).
Flagella and chemotaxis
Genes encoding proteins for the basal body, hook and filament assembly are present in the NICIL-2 genome. However, NICIL-2 lacks genes encoding chemotaxis machinery, apart from one regulatory protein, CheY. The GSB are considered to be non-motile and lack chemotaxis and flagella, while I. album, M. roseus and R. marinus have flagellar and chemotaxis machinery.
Discussion
Microbial community analysis of thermophilic bacterial consortia adapted grow on plant biomass has indicated that the populations enriched in these laboratory cultivations are closely related to organisms (Thermus thermophilus and Rhodothermus marinus) isolated from thermal springs, but which are often found in low abundance because the natural environment is carbon limited. The recovery of the genome of NICIL-2 provides a striking example of the utility of these adapted cultures to provide new insights into populations in natural environments, as the recovery of the NICIL-2 genome provides phylogenetic and metabolic context for an uncultivated lineage that is ubiquitous in thermal environments (Hugenholtz et al., 1998; Nunoura et al., 2005), but for which almost no genomic information was available. The recovered NICIL-2 genome was used to probe metagenomic data from thermal springs and six additional NICIL-2-related genomes were recovered from four distinct environments, five of which clustered with the OPB56 lineage and one of which clustered with the Ignavibacteria. These recovered genomes were almost all at <1% abundance in these binned metagenomes, except for the bin from Nevada Great Boiling Spring (3.2% abundance), which was recovered from an enrichment culture.
The recovered genome of NICIL-2 and related genomes has refined the description of the phylum Chlorobi based on 16S rRNA gene and concatenated marker gene phylogenetic trees. Previous descriptions of the Ignavibacteria based on the physiological and genomic studies of I. album and M. roseum had disagreed on the assignment of the Ignavibacteria as a class in the Chlorobi (Liu et al., 2012a), or as a separate phylum (Podosokorskaya et al., 2013). Both the 16S rRNA gene and protein phylogenetic trees described in this work that include the OPB56 lineage agree with the assignment of the Ignavibacteria as a class in the Chlorobi, not a distinct phylum, since the Ignavibacteria and the Chlorobea are sister clades in both 16S rRNA and multiple protein trees (Figure 2 and 3). The 16S rRNA phylogenetic tree (Figure 2) supports OPB56 as a separate phylum level lineage that branches before the node containing the Chlorobea, Ignavibacteria and the Bacteroidetes. However, a number of recent studies that reconstruct the genome of deeply branching lineages have identified discrepancies between the 16S rRNA gene trees and ribosomal protein-based phylogenies (Castelle et al., 2013; Dodsworth et al., 2013). Concatenated protein trees have been proposed as a more accurate method to determine evolutionary relationships compared to 16S rRNA gene-based trees, despite complications with horizontal gene transfer (Lang et al., 2013). The concatenated ribosomal protein tree (Figure 3a) located the OPB56 lineage at the same node as the GSB and Ignavibacteria, suggesting that the three lineages have a common ancestor that diverged before establishment of the Bacteroidetes. As this observation contradicted the 16S rRNA-based phylogeny, a second protein-based phylogenetic tree was constructed by concatenating 86 single copy genes conserved among the lineages to construct a genome-wide phylogenetic comparison (Soo et al., 2014). This comparison reproduced the topology from the ribosomal tree, providing further support for the affiliation of the Chlorobea, Ignavibacteria and OPB56 as classes in a single phylum. Conserved insertions in DNA polymerase III and alanyl-tRNA-synthetase demonstrated that the GSB were distinct from the other lineages (Supplementary Figures S2 and S3); however, a portion of the insertion into the alanyl-tRNA synthetase sequence was conserved in the Ignavibacteria and NICIL-2 predicted proteins, providing further evidence for the clustering of the Chlorobea, Ignavibacteria and OPB56.
The metabolic potential of NICIL-2 indicates a close relationship with the Ignavibacteria and the Bacteroidetes, represented by the most deeply branching lineage of the phylum, the family Rhodothermaceae. All of these groups have a chemoheterotrophic lifestyle, with NICIL-2 having the simplest apparent carbon metabolism. Despite its abundance as part of a biomass-deconstructing consortium, it has limited metabolic potential for the deconstruction of complex biomass. Therefore, it is likely a secondary consumer of simple sugars or amino acids produced during biomass deconstruction by community members with the ability to hydrolyze insoluble polysaccharides (Eichorst et al., 2013). Recovery of additional OPB56-related genomes from natural samples supports the inference that the metabolic potential for deconstructing complex biomass is not present in this lineage. Therefore, the members of the OPB56 lineage likely are heterotrophs broadly distributed in thermal environments that metabolize small organic molecules. In contrast, R. marinus (Rhodothermaceae) and M. roseus (Ignavibacteria) have an extensive set of genes encoding for plant biomass deconstruction. However, this metabolic potential does not appear indicative of the lineage with which they affiliate, as S. ruber (Rhodothermaceae) and I. album (Ignavibacteria) have more limited metabolic potential for biomass deconstruction (Supplementary Figure S5).
Electron transport chains are often highly conserved and provide a basis to compare the lineages (Li and Graur, 1991). Concatenated protein trees of Complex I demonstrated that the electron transport chains of NICIL-2 are more closely related to the Rhodothermaceae than the Ignavibacteria, emphasizing the link between these two deep branching groups assigned to different phyla. The six additional NICIL-2-related genomes had Complex I subunits whose concatenated sequences clustered with NICIL-2 and were more closely related to the Rhodothermaceae. Complex I from the Ignavibacteria is located on the same branch as the GSB, confirming the closer phylogenetic linkage between the Ignavibacteria and the Chlorobea found in the 16S rRNA and protein phylogenetic trees. In contrast to Complex I, the ACIII sequences demonstrated a more complex relationship. The presence of homologs of ACIII in the Rhodothermaceae, Ignavibacteria and NICIL-2 suggests that it was present in a common ancestor and was lost by the GSB during the transition from a faculative to strict anaerobic lifestyle demonstrated by the presence of ACIII in ‘Ca. T. aerophilum’.
The expansion of the phylum Chlorobi to include heterotrophic lineages such as the Ignavibacteria (Liu et al., 2012a) and OPB56 and the observation of close phylogenetic relationships between proteins encoded in the NICIL-2 genome and the Rhodothermaceae provide an opportunity to rethink the classical division between the phyla Bacteroidetes and Chlorobi (Hugenholtz et al., 1998; Gupta, 2004). This division was based on the pronounced phenotypic differences between cultured Bacteroidetes and GSB isolates. Based on recent work, the GSB may represent a specific adaptation to environmental conditions, including acquisition of genes for photosynthesis by horizontal gene transfer (Gupta, 2010), that arose from a heterotrophic lineage with more phenotypic similarity to the Bacteroidetes. The targeted recovery of genomes from uncultivated organisms related to the NICIL-2 provides a promising route to build a more detailed evolutionary model linking the Bacteroidetes and the Chlorobi.
Accession codes
References
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH . (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538.
Allgaier M, Reddy A, Park JI, Ivanova N, D'haeseleer P, Lowry S et al. (2010). Targeted discovery of glycoside hydrolases from a switchgrass-adapted compost community. PLoS One 5: e8812.
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402.
Arakawa C, Kuratsu M, Furihata K, Hiratsuka T, Itoh N, Seto H et al. (2011). Diversity of the early steps of the futalosine pathway. Antimicrob Agents Chemother 55: 913–916.
Bentley R, Meganathan R . (1982). Biosynthesis of vitamin K (menaquinone) in bacteria. Microbiol Rev 46: 241–280.
Buchanan BB, Arnon DI . (1990). A reverse Krebs cycle in photosynthesis—consensus at last. Photosynth Res 24: 47–53.
Castelle CJ, Hug LA, Wrighton KC, Thomas BC, Williams KH, Wu D et al. (2013). Extraordinary phylogenetic diversity and metabolic versatility in aquifer sediment. Nat Commun 4: 2120.
Chan LK, Weber TS, Morgan-Kiss RM, Hanson TE . (2008). A genomic region required for phototrophic thiosulfate oxidation in the green sulfur bacterium Chlorobium tepidum (syn. Chlorobaculum tepidum. Microbiology 154: 818–829.
D'haeseleer P, Gladden JM, Allgaier M, Chain PS, Tringe SG, Malfatti SA et al. (2013). Proteogenomic analysis of a thermophilic bacterial consortium adapted to deconstruct switchgrass. PLoS One 8: e68465.
Dibble DC, Li C, Sun L, George A, Cheng A, Cetinkol OP et al. (2011). A facile method for the recovery of ionic liquid and lignin from biomass pretreatment. Green Chem 13: 3255–3264.
Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP et al. (2009). Community-wide analysis of microbial genome sequence signatures. Genome Biol 10: R85.
Dodsworth JA, Blainey PC, Murugapiran SK, Swingley WD, Ross CA, Tringe SG et al. (2013). Single-cell and metagenomic analyses indicate a fermentative and saccharolytic lifestyle for members of the OP9 lineage. Nat Commun 4: 1854.
Dupont CL, Rusch DB, Yooseph S, Lombardo MJ, Richter RA, Valas R et al. (2012). Genomic insights to SAR86, an abundant and uncultivated marine bacterial lineage. ISME J 6: 1186–1199.
Edgar RC . (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
Eichorst SA, Varanasi P, Stavila V, Zemla M, Auer M, Singh S et al. (2013). Community dynamics of cellulose-adapted thermophilic bacterial consortia. Environ Microbiol 15: 2573–2587.
Eichorst SA, Joshua C, Sathitsuksanoh N, Singh S, Simmons BA, Singer SW . (2014). Substrate-specific development of thermophilic bacterial consortia by using chemically pretreated switchgrass. Appl Environ Microbiol 80: 7423–7432.
Eisen JA, Nelson KE, Paulsen IT, Heidelberg JF, Wu M, Dodson RJ et al. (2002). The complete genome sequence of Chlorobium tepidum TLS, a photosynthetic, anaerobic, green-sulfur bacterium. Proc Natl Acad Sci USA 99: 9509–9514.
Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR et al. (2014). Pfam: the protein families database. Nucleic Acids Res 42: D222–D230.
Fouke BW, Bonheyo GT, Sanzenbacher B, Frias-Lopez J . (2003). Partitioning of bacterial communities between travertine depositional facies at Mammoth Hot Springs, Yellowstone National Park, USA. Can J Earth Sci 40: 1531–1548.
Frigaard NU, Dahl C . (2009). Sulfur metabolism in phototrophic sulfur bacteria. Adv Microb Physiol 54: 103–200.
Gao X, Xin Y, Blankenship RE . (2009). Enzymatic activity of the alternative complex III as a menaquinol:auracyanin oxidoreductase in the electron transfer chain of Chloroflexus aurantiacus. FEBS Lett 583: 3275–3279.
Gao X, Majumder EW, Kang Y, Yue H, Blankenship RE . (2013). Functional analysis and expression of the mono-heme containing cytochrome c subunit of Alternative Complex III in Chloroflexus aurantiacus. Arch Biochem Biophys 535: 197–204.
Gladden JM, Allgaier M, Miller CS, Hazen TC, VanderGheynst JS, Hugenholtz P et al. (2011). Glycoside hydrolase activities of thermophilic bacterial consortia adapted to switchgrass. Appl Environ Microbiol 77: 5804–5812.
Gregersen LH, Bryant DA, Frigaard NU . (2011). Mechanisms and evolution of oxidative sulfur metabolism in green sulfur bacteria. Front Microbiol 2: 116.
Gupta RS . (2004). The phylogeny and signature sequences characteristics of Fibrobacteres, Chlorobi, and Bacteroidetes. Crit Rev Microbiol 30: 123–143.
Gupta RS, Lorenzini E . (2007). Phylogeny and molecular signatures (conserved proteins and indels) that are specific for the Bacteroidetes and Chlorobi species. BMC Evol Biol 7: 71.
Gupta RS . (2010). Molecular signatures for the main phyla of photosynthetic bacteria and their subgroups. Photosynth Res 104: 357–372.
Heising S, Richter L, Ludwig W, Schink B . (1999). Chlorobium ferrooxidans sp. nov., a phototrophic green sulfur bacterium that oxidizes ferrous iron in coculture with a ‘Geospirillum’ sp. strain. Arch Microbiol 172: 116–124.
Hess M, Sczyrba A, Egan R, Kim TW, Chokhawala H, Schroth G et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331: 463–467.
Hugenholtz P, Pitulle C, Hershberger KL, Pace NR . (1998). Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol 180: 366–376.
Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC . (2011). Integrative analysis of environmental sequences using MEGAN4. Genome Res 21: 1552–1560.
Imhoff JF . (2003). Phylogenetic taxonomy of the family Chlorobiaceae on the basis of 16S rRNA and fmo (Fenna-Matthews-Olson protein) gene sequences. Int J Syst Evol Microbiol 53: 941–951.
Kadnikov VV, Mardanov AV, Podosokorskaya OA, Gavrilov SN, Kublanov IV, Beletsky AV et al. (2013). Genomic analysis of Melioribacter roseus, facultatively anaerobic organotrophic bacterium representing a novel deep lineage within Bacteriodetes/Chlorobi Group. Plos One 8: e53047.
Lang JM, Darling AE, Eisen JA . (2013). Phylogeny of bacterial and archaeal genomes using conserved genes: supertrees and supermatrices. PLoS One 8: e62510.
Li H, Durbin R . (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760.
Li W-H, Graur D . (1991) Fundamentals of Molecular Evolution vol. 48. Sinauer Associates: Sunderland, MA.
Liu Z, Frigaard NU, Vogl K, Iino T, Ohkuma M, Overmann J et al. (2012a). Complete genome of Ignavibacterium album, a metabolically versatile, flagellated, facultative anaerobe from the phylum Chlorobi. Front Microbiol 3: 185.
Liu Z, Klatt CG, Ludwig M, Rusch DB, Jensen SI, Kuhl M et al. (2012b). 'Candidatus Thermochlorobacter aerophilum:' an aerobic chlorophotoheterotrophic member of the phylum Chlorobi defined by metagenomics and metatranscriptomics. ISME J 6: 1869–1882.
Liu Z, Mueller J, Li T, Alvey RM, Vogl K, Frigaard N-U et al. (2013). Genomic analysis reveals key aspects of prokaryotic symbiosis in the phototrophic consortium ‘Chlorochromatium aggregatum’. Genome Biol 14: R127.
Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B . (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42: D490–D495.
Mackelprang R, Waldrop MP, DeAngelis KM, David MM, Chavarria KL, Blazewicz SJ et al. (2011). Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 480: 368–371.
Markowitz VM, Chen IM, Chu K, Szeto E, Palaniappan K, Pillay M et al. (2014). IMG/M 4 version of the integrated metagenome comparative analysis system. Nucleic Acids Res 42: D568–D573.
Mistry J, Finn RD, Eddy SR, Bateman A, Punta M . (2013). Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res 41: e121.
Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M . (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35: W182–W185.
Nolan M, Tindall BJ, Pomrenke H, Lapidus A, Copeland A, Glavina Del Rio T et al. (2009). Complete genome sequence of Rhodothermus marinus type strain (R-10). Stand Genomic Sci 1: 283–290.
Nunoura T, Hirayama H, Takami H, Oida H, Nishi S, Shimamura S et al. (2005). Genetic and functional properties of uncultivated thermophilic crenarchaeotes from a subsurface gold mine as revealed by analysis of genome fragments. Environ Microbiol 7: 1967–1984.
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res 42: D206–D214.
Overmann J . (2008) ‘Ecology of phototrophic sulfur bacteria.’ Sulfur Metabolism in Phototrophic Organisms Vol 27 Springer: Netherlands, pp 375–396.
Park JI, Steen EJ, Burd H, Evans SS, Redding-Johnson AM, Batth T et al. (2012). A thermophilic ionic liquid-tolerant cellulase cocktail for the production of cellulosic biofuels. PLoS One 7: e37010.
Pereira MM, Refojo PN, Hreggvidsson GO, Hjorleifsdottir S, Teixeira M . (2007). The alternative complex III from Rhodothermus marinus—a prototype of a new family of quinol:electron acceptor oxidoreductases. FEBS Lett 581: 4831–4835.
Podosokorskaya OA, Kadnikov VV, Gavrilov SN, Mardanov AV, Merkel AY, Karnachuk OV et al. (2013). Characterization of Melioribacter roseus gen. nov., sp nov., a novel facultatively anaerobic thermophilic cellulolytic bacterium from the class Ignavibacteria, and a proposal of a novel bacterial phylum Ignavibacteriae. Environ Microbiol 15: 1759–1771.
Rappe MS, Giovannoni SJ . (2003). The uncultured microbial majority. Annu Rev Microbiol 57: 369–394.
Refojo PN, Ribeiro MA, Calisto F, Teixeira M, Pereira MM . (2013). Structural composition of alternative complex III: variations on the same theme. Biochim Biophys Acta 1827: 1378–1382.
Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437.
Rodriguez J, Hiras J, Hanson TE . (2011). Sulfite oxidation in Chlorobaculum tepidum. Front Microbiol 2: 112.
Ross KA, Feazel LM, Robertson CE, Fathepure BZ, Wright KE, Turk-Macleod RM et al. (2012). Phototrophic phylotypes dominate mesothermal microbial mats associated with hot springs in Yellowstone National Park. Microb Ecol 64: 162–170.
Sharon I, Morowitz MJ, Thomas BC, Costello EK, Relman DA, Banfield JF . (2013). Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res 23: 111–120.
Soo RM, Skennerton CT, Sekiguchi Y, Imelfort M, Paech SJ, Dennis PG et al. (2014). An expanded genomic representation of the phylum Cyanobacteria. Genome Biol Evol 6: 1031–1045.
Talavera G, Castresana J . (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56: 564–577.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S . (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739.
Thiel V, Hamilton TL, Tomsho LP, Burhans R, Gay SE, Ramaley RF et al. (2014). Draft genome sequence of the moderately thermophilic bacterium Schleiferia thermophila strain Yellowstone (Bacteroidetes). Genome Announc 2: e00860–00814.
Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM et al. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428: 37–43.
Wahlund TM, Madigan MT . (1993). Nitrogen-Fixation by the Thermophilic Green Sulfur Bacterium Chlorobium tepidum. J Bacteriol 175: 474–478.
Wahlund TM, Tabita FR . (1997). The reductive tricarboxylic acid cycle of carbon dioxide assimilation: Initial studies and purification of ATP-citrate lyase from the green sulfur bacterium Chlorobium tepidum. J Bacteriol 179: 4859–4867.
Woese C, Stackebrandt E, Macke T, Fox G . (1985). A phylogenetic definition of the major eubacterial taxa. Syst Appl Microbiol 6: 143–151.
Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC et al. (2012). Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337: 1661–1665.
Wu Y, Hsu Y, Tringe SG, Simmons BA, Singer SW . (2014). MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2: 26.
Yanyushin MF, del Rosario MC, Brune DC, Blankenship RE . (2005). New class of bacterial membrane oxidoreductases. Biochemistry 44: 10037–10045.
Yin Y, Mao X, Yang J, Chen X, Mao F, Xu Y . (2012). dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40: W445–W451.
Acknowledgements
This work was performed as part of the DOE Joint BioEnergy Institute (http://www.jbei.org) supported by the US Department of Energy, Office of Science, Office of Biological and Environmental Research, through contract DE-AC02-05CH11231 between Lawrence Berkeley National Laboratory and the US Department of Energy. Metagenomic sequencing was conducted by the Joint Genome Institute, which is supported by the Office of Science of the US Department of Energy under Contract No. DE-AC02-05CH11231. We would like to thank Susannah Tringe, Tijana Glavina Del Rio and Stephanie Malfatti of the Joint Genome Institute for their assistance in obtaining and processing metagenomic sequencing data. We would also like to thank Professor Thomas E Hanson (University of Delaware) for helpful comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on The ISME Journal website
Supplementary information
Rights and permissions
About this article

Cite this article
Hiras, J., Wu, YW., Eichorst, S. et al. Refining the phylum Chlorobi by resolving the phylogeny and metabolic potential of the representative of a deeply branching, uncultivated lineage. ISME J 10, 833–845 (2016). https://doi.org/10.1038/ismej.2015.158
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ismej.2015.158
This article is cited by
-
Microbial community composition, dynamics, and biogeochemistry during the start-up of a partial nitritation-anammox pathway in an upflow reactor
Sustainable Environment Research (2022)
-
Activity-based, genome-resolved metagenomics uncovers key populations and pathways involved in subsurface conversions of coal to methane
The ISME Journal (2022)
-
Diversity assessment of photosynthesizers: comparative analysis of pre-cultivated and natural microbiome of sediments from Cerrado biome in Maranhão, Brazil
Environmental Science and Pollution Research (2022)
-
Multidomain ribosomal protein trees and the planctobacterial origin of neomura (eukaryotes, archaebacteria)
Protoplasma (2020)
-
Biodegradation of natural rubber and deproteinized natural rubber by enrichment bacterial consortia
Biodegradation (2020)
