
Abstract
The widespread occurrence of sulfate-reducing microorganisms (SRMs) in temporarily oxic/hypoxic aquatic environments indicates an intriguing possibility that SRMs can prevail in constantly oxic/hypoxic terrestrial sulfate-rich environments. However, little attention has been given to this possibility, leading to an incomplete understanding of microorganisms driving the terrestrial part of the global sulfur (S) cycle. In this study, genome-centric metagenomics and metatranscriptomics were employed to explore the diversity, metabolic potential, and gene expression profile of SRMs in a revegetated acidic mine wasteland under constantly oxic/hypoxic conditions. We recovered 16 medium- to high-quality metagenome-assembled genomes (MAGs) containing reductive dsrAB. Among them, 12 and four MAGs belonged to Acidobacteria and Deltaproteobacteria, respectively, harboring three new SRM genera. Comparative genomic analysis based on seven high-quality MAGs (completeness >90% and contamination <10%; including six acidobacterial and one deltaproteobacterial) and genomes of three additional cultured model species showed that Acidobacteria-related SRMs had more genes encoding glycoside hydrolases, oxygen-tolerant hydrogenases, and cytochrome c oxidases than Deltaproteobacteria-related SRMs. The opposite pattern was observed for genes encoding superoxide reductases and thioredoxin peroxidases. Using VirSorter, viral genome sequences were found in five of the 16 MAGs and in all three cultured model species. These prophages encoded enzymes involved in glycoside hydrolysis and antioxidation in their hosts. Moreover, metatranscriptomic analysis revealed that 15 of the 16 SRMs reported here were active in situ. An acidobacterial MAG containing a prophage dominated the SRM transcripts, expressing a large number of genes involved in its response to oxidative stress and competition for organic matter.
Similar content being viewed by others


Introduction
Sulfate-reducing microorganisms (SRMs) are characterized by their ability to grow with energy derived from the reduction of sulfate to sulfide1,2. The canonical dissimilatory sulfate reduction pathway in SRMs is driven by a set of enzymes3, including sulfate adenylyltransferase (Sat), adenylyl sulfate reductase (AprBA), and dissimilatory sulfite reductase (DsrAB). More specifically, Sat and AprBA, encoded by sat and aprBA, cooperate to complete the reduction of sulfate to sulfite2. Reductive DsrAB, encoded by reductive dsrAB, interacts with DsrC to reduce sulfite to sulfide4,5, a rate-limiting step in the biogeochemical cycle of S on Earth6,7.
Due to the difficulty in isolating pure cultures of SRMs8, the functional genes aprBA and reductive dsrAB have been widely employed to explore SRM diversity in the environment9,10,11,12,13. A striking example is the work of Vigneron et al.13. The authors found 167,397 different species-level dsrB OTUs affiliated with 47 different families in 14 different ecosystems. Among these OTUs, ~99% were previously not detected, greatly improving our knowledge of the global species-level biodiversity of SRMs.
Recently, a new trend in SRM research has been the application of genome-centric metagenomics14,15,16,17,18. One of the most important advantages of this approach lies in the recovery of near-complete genomes representing species-level microorganisms of interest19. This advantage not only allows the identification of previously uncultured microorganisms of interest but also provides insights into the metabolic potentials of microorganisms of interest20,21,22, although some findings from metagenomics studies cannot be definitely confirmed until pure cultures of microorganisms of interest are obtained. Nonetheless, recent studies on MAGs containing reductive dsrAB from environments have revealed that: (1) eight prokaryotic phyla that were not previously reported to have SRMs harbor the canonical functional genes required for dissimilatory sulfate reduction14; (2) two acidobacterial MAGs encoding reductive DsrAB but not Sat and AprBA contain sulfite-producing enzyme genes that allow them to use organosulfonates as growth substrates15; and (3) viruses can infect SRMs in wetland sediments and thus likely affect the functions of their hosts in previously unknown ways16. Furthermore, although very rare, genome-centric metatranscriptomic evidence suggests that previously unknown Acidobacteria-related SRMs play an important role in sulfate reduction in anoxic peat soils15.
However, little is known about SRMs in terrestrial environments2. This situation represents an incomplete understanding of microorganisms responsible for the terrestrial part of the global S cycle, as the geographic distribution of sulfate-rich soils is not restricted to aquatic environments23. Many natural processes (e.g., prolonged droughts) and anthropogenic interventions (including mining operations) can lead to the distribution of sulfate-rich soils in terrestrial environments23. A major distinction between terrestrial and aquatic sulfate-rich soils lies in the constantly oxic/hypoxic conditions associated with the former, although oxic/hypoxic conditions can temporarily exist in the latter24. Such discrepancies, however, should not preclude the occurrence and activity of SRMs in terrestrial sulfate-rich soils.
The functioning of SRMs in temporarily oxic/hypoxic aquatic environments has long been recognized25,26. Several cultivated model SRMs in Deltaproteobacteria are known to possess a variety of enzymes enabling them to tolerate oxidative stress24. A recent study obtained the pure SRM cultures (i.e., Desulfovibrio vulgaris strains) that could grow using energy derived from oxygen reduction27. In contrast, the existing literature contains only a few lines of evidence demonstrating the occurrence or activity of SRMs in terrestrial sulfate-rich soils under constantly oxic/hypoxic conditions28. A previous study showed that a majority of potential SRMs in oxic/hypoxic mine tailings (i.e., the materials left after extraction and beneficiation of ores) were affiliated with Firmicutes and Deltaproteobacteria29. In addition, two acidobacterial MAGs retrieved from an acidic mine site were found to encode canonical enzymes required for sulfate reduction14. Nonetheless, no information on the transcriptomes of SRMs under constantly oxic/hypoxic conditions has been reported14.
Given the abovementioned information, we hypothesized that diverse SRMs can be alive in sulfate-rich mine wastelands under constantly oxic/hypoxic conditions but their survival strategies likely differ considerably between lineages. To test our hypothesis, we employed genome-centric metagenomics and metatranscriptomics to characterize SRMs in a revegetated acidic mine wasteland. Before revegetation, the wasteland (a mine tailings pond; pH ~2.5) was abandoned and drained for eight years. A promising remediation technology termed ‘phytostabilization’ was used to revegetate the wasteland30, thereby facilitating in situ stabilization of tailings and metal contaminants. Approximately 4000 m2 of vegetation was established on the wasteland, and it increasingly flourished as time progressed30. This revegetated mine wasteland was selected because: (1) it was a representative case illustrating the effectiveness of phytostabilization for remediating sulfate-rich mine wastelands distributed globally; and (2) it consisted of different habitats with soil oxidation-reduction potential (Eh) values varying from ~180–680 mV31, a representative Eh range encountered in terrestrial environments under constantly oxic/hypoxic conditions32.
Results
MAGs harboring reductive dsrAB
We identified 50 DsrAB protein sequences in 982 medium- to high-quality MAGs (completeness >50% and contamination <10%) from 18 metagenomes from the revegetated acid mine wasteland, which were published elsewhere31. Sixteen of these sequences belonged to the reductive bacterial-type DsrAB family (Supplementary Fig. 1). Accordingly, 16 reductive dsrAB-containing MAGs were retrieved, with 12 from Acidobacteria and four from Deltaproteobacteria (Fig. 1 and Supplementary Table 1).

Sixteen medium- to high-quality MAGs (completeness >50% and contamination <10%) were included in the analysis. The functions of the MAGs in sulfate reduction were predicted according to the presence and/or absence of key genes for the pathway. A representative organization of dissimilatory sulfate reduction genes on the MAGs belonging to Acidobacteria and Deltaproteobacteria is displayed, respectively. Tree scale bar = 1. Additional details are provided in Supplementary Tables 1 and 2.
The 12 acidobacterial MAGs were all affiliated with subdivision 1 of Acidobacteria (Supplementary Fig. 2). Among them, three (i.e., Ac.bin4, Ac.bin9, and Ac.bin6) formed a monophyletic clade and had an average amino acid identity (AAI) of 63% with its closest relative (Granulicella tundricola MP5ACTX9)33. Similarly, Ac.bin5 formed a monophyletic clade and had AAIs of 56–62% with its closest relatives. The four acidobacterial MAGs mentioned above likely represented two new SRM genera, given that no SRMs from Acidobacteria have been successfully cultivated and that the currently known acidobacterial MAGs containing dsrAB15 were not affiliated with the genera represented by the four MAGs (Supplementary Fig. 2).
Three out of the four reductive dsrAB-containing deltaproteobacterial MAGs were affiliated with the well-known SRM genus Desulfovibrio (Supplementary Table 1). However, the remaining one (i.e., De.bin4) from the family Syntrophobacteraceae formed a monophyletic clade and could not be assigned to a specific genus with references available (Supplementary Fig. 3). Specifically, De.bin4 had AAIs of 54–60% with its closest relatives. Therefore, we inferred that it belonged to a new genus.
The Dsr operon structures of Acidobacteria were different from those of Deltaproteobacteria (Fig. 1). Multiple alignments of the DsrD and DsrT sequences with published references confirmed the highly conserved residues (Supplementary Figs. 4 and 5), indicating that these proteins were likely functional. According to the common patterns for determining the direction of dissimilatory S metabolism for uncultivated microorganisms14, 11 MAGs (eight from Acidobacteria and three from Deltaproteobacteria) in this study encoded a complete pathway for the reduction of sulfate to sulfide (Fig. 1 and Supplementary Table 2). Notably, seven high-quality MAGs (completeness >90% and contamination <10%) of SRMs (six acidobacterial and one deltaproteobacterial, Supplementary Table 1) were obtained and thus further analyzed for their metabolism. For comparison, the complete genomes of one aerobic acidobacterial species (i.e., Terracidiphilus gabretensis S55, non-SRM)34, Desulfovibrio vulgaris Hildenborough (an oxygen-tolerant cultured model SRM), and Desulfococcus multivorans DSM 2059 (an oxygen-sensitive cultured model SRM)35 were also chosen for functional analysis.
Glycoside hydrolysis of SRMs
Seventy-one glycoside hydrolase (GH) families were encoded by the 10 focal genomes (including six high-quality acidobacterial MAGs and one high-quality deltaproteobacterial MAG, as well as the complete genomes of one cultured acidobacterial species and two cultured deltaproteobacterial SRMs; Supplementary Table 3). Among them, all but one family (i.e., GH50) was not found in the seven acidobacterial genomes. In contrast, the three deltaproteobacterial genomes encoded only 10 GH families. Judging from the average number of genes encoding a given GH family per genome, GH3, GH13, GH23, GH2, GH31, GH29, GH28, GH27, GH92, and GH35 were the 10 most abundant genes across all 10 focal genomes (Fig. 2). Except for GH23, these abundant GH families were largely represented by acidobacterial genomes. A striking example was GH3, which was encoded by 9–13 genes in each acidobacterial genome but by only one gene per deltaproteobacterial genome (Fig. 2).

Seven high-quality MAGs (completeness >90% and contamination <10%) of SRMs were included in the analysis. For comparison, the complete genomes of one aerobic acidobacterial species (i.e., Terracidiphilus gabretensis S55, non-SRM), Desulfovibrio vulgaris Hildenborough (an oxygen-tolerant cultured model SRM), and Desulfococcus multivorans DSM 2059 (an oxygen-sensitive cultured model SRM) were chosen. The names of acidobacterial genomes are in blue. The 10 most abundant glycoside hydrolase families across the genomes are shown. Those hydrogenase subgroups that are known to be involved in sulfate reduction or to be oxygen tolerant are listed. MCP methyl-accepting chemotaxis protein, BCP bacterioferritin comigratory protein, Cbo cytochrome bd oxygen reductase, Cco cytochrome c oxidase, CcPx cytochrome c peroxidase, SOD superoxide dismutase, CysK cysteine synthase, GPX glutathione peroxidase, NPX NADH peroxidase, ROO rubredoxin-oxygen oxidoreductase, SOR superoxide reductase, TPX thioredoxin peroxidase. Additional details are presented in Supplementary Tables 3–8.
Hydrogen metabolism of SRMs
Genes encoding eight groups of hydrogenases (including groups A1 and A2 of [FeFe]-hydrogenase and groups 1a, 1b, 1d, 3d, 4c, and 4e of [NiFe]-hydrogenase) were identified on the 10 focal genomes (Supplementary Table 4). Among them, [FeFe]-hydrogenase was encoded only by D. vulgaris. These results were consistent with those of Hausmann et al.15, who showed that MAGs of Acidobacteria-related SRMs harbored genes encoding groups 1 (excluding 1 h), 3, and 4 of [NiFe]-hydrogenase. Examination of individual genomes showed that they differed considerably in the total number of hydrogenase genes (Supplementary Table 4). The genome of D. vulgaris contained up to seven hydrogenase genes, while three acidobacterial genomes (i.e., Ac.bin6, Ac.bin4 and T. gabretensis) lacked such genes. Despite this, seven out of the eight genes encoding oxygen-tolerant hydrogenases (i.e., groups 1d and 3d of [NiFe]-hydrogenase36) were identified in the remaining four acidobacterial genomes (Fig. 2).
Respiratory chain of SRMs
All 10 selected genomes encoded the major components of the respiratory chain (Supplementary Table 5). Specifically, the (near) complete operons for NADH dehydrogenase 1 (lacking in D. vulgaris and D. multivorans), NADH dehydrogenase 2 (lacking in Ac.bin6 and Ac.bin4), succinate dehydrogenase, quinol–cytochrome-c reductase, high-affinity terminal oxidase (lacking in T. gabretensis), low-affinity terminal oxidase (lacking in D. multivorans), and F-type ATP synthase were detected. Remarkably, bd-type terminal oxidase was encoded by all the focal genomes except that of T. gabretensis, while cbb3-type terminal oxidase was detected in only an acidobacterial MAG (i.e., Ac.bin3, Supplementary Table 5).
Methyl-accepting chemotaxis protein system of SRMs
Compared with the seven acidobacterial genomes, two Desulfovibrio-related genomes (i.e., De.bin1 and D. vulgaris) encoded many more methyl-accepting chemotaxis protein (MCPs; Fig. 2 and Supplementary Table 6), which showed a high level of sequence similarity between these two SRMs (Supplementary Fig. 6). No MCP genes were detected in Ac.bin3 and D. multivorans. Among the seven known MCP classes37, five were recorded here (including classes Ia, IIIm, IIIc, IVa, and IVb). Although the majority of the MCPs encoded by the two Desulfovibrio-related genomes belonged to class Ia (including clusters I and II; Supplementary Table 7), the acidobacterial MCPs were mainly from classes IVa and IVb. Remarkably, among the 10 focal genomes, Ac.bin5 was the only one that harbored both class Ia (cluster I) and class IVa MCP genes (Supplementary Table 7).
A complete set of genes encoding core chemotaxis signaling complexes (i.e., CheB, CheR, CheW, CheA, and CheY)38 were detected in almost all acidobacterial genomes (except Ac.bin3) and the two Desulfovibrio-related genomes (Fig. 2 and Supplementary Table 6). In addition, the che operon structure in De.bin1 was the same as that of cheA3 in D. vulgaris (Supplementary Fig. 7), suggesting that it had the ability to sense sulfate as an electron acceptor and lactate as an electron donor39.
Flagellar system of SRMs
A complete set of 24 core flagellar genes40 was identified in five acidobacterial genomes (i.e., Ac.bin1, Ac.bin2, Ac.bin3, Ac.bin5, and T. gabretensis) and two Desulfovibrio-related genomes (i.e., De.bin1 and D. vulgaris; Fig. 2 and Supplementary Table 6). Genes encoding highly conserved components of the type IV pilus (e.g., pilA) were identified in Ac.bin4 and Ac.bin6 (Supplementary Table 6), indicating that these two SRMs could move towards chemoattractants using pili-based “twitching” motility41. These results, together with those for the MCP system, suggested that six genomes (i.e., Ac.bin1, Ac.bin2, Ac.bin5, T. gabretensis, De.bin1, and D. vulgaris) had potential to utilize flagellum-driven chemotaxis to sense surrounding chemoattractants and relocate themselves towards favorable microenvironments.
Antioxidative enzymes of SRMs
Among the four known enzymes involved in oxygen reduction by SRMs24, only cytochrome bd oxygen reductase (Cbo, EC 7.1.1.7) was encoded by all 10 focal genomes except T. gabretensis (Fig. 2 and Supplementary Table 8). The other three enzymes showed two contrasting patterns: (1) [Fe] hydrogenase (EC 1.12.7.2) and rubredoxin-oxygen oxireductase (ROO) were encoded largely by deltaproteobacterial genomes; and (2) cytochrome c oxidase (Cco, EC 7.1.1.9) occurred mainly in acidobacterial genomes (Fig. 2). Similarly, two opposite trends were observed for the two major enzymes responsible for eliminating superoxide anion radicals:24 (1) all the investigated genomes contained at least one SOD (EC 1.15.1.1) gene, although the type of SOD differed among genomes; and (2) superoxide reductase (SOR, EC 1.15.1.2) genes were present only in deltaproteobacterial genomes (Fig. 2). Note that the majority of the acidobacterial genomes lacked genes encoding catalase (EC 1.11.1.6) and thioredoxin peroxidase (Tpx, EC 1.11.1.15), while they harbored more genes encoding thioredoxin-dependent peroxiredoxin (BCP, EC 1.11.1.24), cysteine synthase (CysK, EC 2.5.1.47) and glutathione peroxidase (GPX, EC 1.11.1.9) than deltaproteobacterial genomes.
Viruses of SRMs
Among the 16 medium- and high-quality MAGs containing reductive dsrAB, five acidobacterial MAGs were found to contain a total of six prophages (with genome sizes varying from 14.0 to 58.2 kb; Fig. 3 and Supplementary Table 9), while no viral sequences were detected in the four deltaproteobacterial MAGs. One, seven, and three prophages were found in T. gabretensis, D. vulgaris, and D. multivorans, respectively, which was not in complete agreement with previous findings showing that one, eight, and no prophages were identified in these three species, respectively33,42,43. This discrepancy likely resulted from the utilization of a more sensitive and accurate viral prediction method (VirSorter;44 see Methods section for details) in this study. Notably, Pfam annotations revealed that 12 out of the 17 prophages identified here harbored at least one virion-associated gene (Supplementary Table 9), suggesting that these prophages still had the genetic potential to complete a lytic cycle33.

The 16 medium- to high-quality MAGs of SRMs were included in the analysis. For comparison, the complete genomes of T. gabretensis, D. vulgaris and D. multivorans were chosen. The cell shapes of the SRMs were drawn based on information from the literature and from our analysis (e.g., the presence or absence of core genes encoding the flagellum). The predicted genome sizes of SRMs and their viruses are shown. Increasing abundances of the SRMs are indicated by darker green colors. The abundance was calculated as the normalized mean coverage depth. Whenever applicable, viral shapes were drawn based on taxonomic information from the signature genes detected in the viral scaffolds. Otherwise, viruses are indicated by hexagons. The hexagons associated with a small rectangle represent tailed viruses. The number (i.e., 1 or 2) within a given virus represents a viral cluster, from which the virus was derived. That is, those hexagons filled with the same number are affiliated with the same viral cluster. Singletons represent new viruses. Increasing abundances of the acidobacterial viruses are indicated by darker red colors. Due to the lack of abundance information, the three cultured species and their viruses are shown in blue and yellow, respectively. Additional details are presented in Supplementary Table 9.
Nine out of the 17 identified prophages could not be clustered with known isolated viruses or those identified in publicly available microbial genomes or metagenomes using a gene content-based classification (genus-level grouping)45, although half of them could be tentatively assigned to the order Caudovirales (Supplementary Table 9). Specifically, five acidobacterial prophages formed an exclusive cluster (named Cluster 1), while the remaining two acidobacterial prophages were not closely related to any previously sequenced bacteriophages at the nucleotide level (referred to as singletons46; Supplementary Table 9 and Fig. 3). Similarly, among the prophages of D. vulgaris and D. multivorans, one was affiliated with the Myoviridae family, three were clustered exclusively (named Cluster 2), and the remaining six were singletons. In addition, the abundance of the prophages targeting acidobacterial SRMs in our study site was positively correlated with host abundance (r = 0.93, P < 0.01; Supplementary Fig. 8).
Roles of viruses in glycoside hydrolysis of SRMs
Three genes encoding GHs were recovered from viral scaffolds, which were further predicted via three-dimensional protein structural modeling (Fig. 4 and Supplementary Table 10). Among them, one was from the virus infecting Ac.bin3 and encoded D-4,5-unsaturated β-glucuronyl hydrolase (EC 3.2.1.172), which is able to release rhamnose from rhamnogalacturonan I oligomers (a major component of the plant cell wall47; Fig. 4). The remaining two genes were identified on the viral scaffolds D. vulgaris.2 and D. vulgaris.5, both of which encoded endochitinase (EC 3.2.1.14). This enzyme can cleave chitin randomly at internal sites, generating soluble low-molecular-mass multimers of N-acetyl-d-glucosamine, such as chitotetraose and chitotriose (Fig. 4)48.

UGL D-4,5-unsaturated β-glucuronyl hydrolase, Endochit endochitinase, MCP methyl-accepting chemotaxis protein, Ni-SOD nickel-containing superoxide dismutase, LBD ligand-binding domain, SD signaling domain, TM transmembrane, HAMP histidine kinase, adenyl cyclase, methyl-accepting chemotaxis protein and phosphatase. Representative substrates of glycoside hydrolases (GHs) encoded by AMGs and the associated simplest products are shown. The cleavage points of the substrates are indicated by red arrows. The computational protein models of GHs, MCPs and Ni-SOD are displayed. Representative ligands of MCPs and the active site of Ni-SOD are shown. *, if the products of GHs or representative ligands of MCPs can be utilized directly by SRMs to reduce sulfate, their names are in blue. Additional details are presented in Supplementary Tables 10 and 11.
Roles of viruses in chemotaxis and antioxidation of SRMs
Three MCPs were encoded by the viral scaffolds D. vulgaris.1 and D. vulgaris.2 (Fig. 4 and Supplementary Table 11). Among them, two belonged to cluster II of class Ia with double cache-like sensor domains, while the other belonged to cluster I of class Ia with a single cache2 domain. According to previous findings49 and the ligands confirmed in model protein structures, lactate and C2/C3 carboxylates (e.g., sodium acetate) could be the ligands for these MCPs (Fig. 4). On the other hand, one gene encoding a Ni-containing SOD was identified on the viral scaffold D. multivorans.2 (Fig. 4 and Supplementary Table 11).
Transcriptomic profile of SRMs
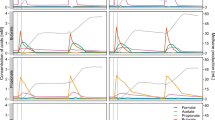
Except for Ac.bin9, all the other MAGs reported in this study were transcriptionally active (Fig. 5a, b), together contributing an average of 0.18% of the total mRNA reads in our metatranscriptomes. Remarkably, a majority of the SRM mRNA reads (51–81%; Fig. 5b) were from Ac.bin5. Specifically, 61–83% of the genes encoded by the SRM were expressed. Furthermore, Ac.bin5 was also an important contributor to the dsrAB transcripts detected in this study, although its relative contribution varied considerably among samples (Fig. 5c, d). The transcript abundance of Ac.bin5 was positively correlated with soil Eh and negatively correlated with soil Fe2+ concentration, while there was a positive relationship between the transcript abundance of dsrAB and soil total carbon content (all r = 0.99, P < 0.03; Supplementary Fig. 9 and Supplementary Table 12).

a Overall transcript abundance of the 16 SRMs. b Relative contribution of individual SRMs to the overall transcript abundance of the 16 SRMs. c Overall transcript abundance of dsrAB of the 16 SRMs. d Relative contribution of individual SRMs to the overall transcript abundance of dsrAB of the 16 SRMs. e Relative expression levels of selected genes of Ac.bin5. Relative expression level was calculated based on the reads per kilobase per million mapped reads (RPKM) of a given selected gene normalized by that of the reference gene gyrA. The gene abbreviations are in accordance with those in Fig. 2. The results are based on three soil samples collected from the unamended layer of the revegetated tailings.
Compared with the housekeeping gene gyrA50, the expression levels of dsrA and dsrB genes of Ac.bin5 were higher, with an average relative expression level (among samples) of 2.5 and 1.8, respectively (Fig. 5e). Note that the relative expression levels of GH genes of Ac.bin5 were extremely high (ranging from 11 to 51; Fig. 5e), with approximately 74–95% of the 129 GH genes of the SRM determined to be active. We found that the genes encoding 1d [NiFe]-hydrogenases, the MCP system (including classes Ia and IVa MCPs), the flagellum system and various antioxidant enzymes (e.g., CbO, Cco, and NPX) of the SRM were also highly expressed, with an average relative expression level ranging from 3.3 to 46 (Fig. 5e).
To further confirm the comparative genomics results, the relative expression levels of selected genes of a representative deltaproteobacterial SRM (i.e., De.bin1, the most dominant high-quality deltaproteobacterial MAG in our metatranscriptomes) were also examined. The genes involving in sulfate reduction and other focal metabolic processes (e.g., glycoside hydrolysis, respiration, denitrification, and antioxidation) were highly expressed as compared to gyrA, with average relative expression levels ranging from 1.4 to 76 (Supplementary Fig. 10 and Supplementary Table 13). Interestingly, the average relative expression level of GH genes was 23-times higher in Ac.bin5 than in De.bin1 (P < 0.05; Supplementary Fig. 10). As to other genes commonly expressed in Ac.bin5 and De.bin1, their relative expression levels showed no significant differences between the two SRMs (P > 0.05). However, the lack of significant differences did not indicate that absolute expression levels of these genes were similar in the two SRMs, because the gyrA of Ac.bin5 in the metatranscriptomes was much more abundant than that of De.bin1 (average RPKM: 13.4 vs 0.17). Although genes encoding BCP and Fe/Mn-SOD were found in both SRMs, only those in Ac.bin5 were expressed (with an average relative expression level of 2.4 and 2.7, respectively). In addition, transcripts of 1d and 3d [NiFe]-hydrogenases and CcO were detected only in Ac.bin5 (Supplementary Fig. 10), which was consistent with our comparative genomics findings that these genes were possessed by Ac.bin5 but not De.bin1 (Fig. 2).
Discussion
We performed a genome-centric metagenomic and metatranscriptomic study that was specifically aimed at characterizing SRMs in a constantly oxic/hypoxic terrestrial environment. The recovery of seven high-quality MAGs of SRMs (six acidobacterial and one deltaproteobacterial) provided us with a particular opportunity to explore the metabolic potentials of these SRMs and their viruses, with emphasis on a comparison between Acidobacteria and Deltaproteobacteria. To this end, the genomes of one aerobic acidobacterial species (i.e., T. gabretensis34) and two cultured deltaproteobacterial model SRMs with contrasting tolerance to oxygen stress (i.e., the oxygen-tolerant D. vulgaris and oxygen-sensitive D. multivorans35) were also included for comparison. We focused on genes encoding enzymes responsible for energy gain (including GHs and hydrogenases1,2) and antioxidation (including antioxidant and chemotaxis enzymes24) because the studied mine wasteland was characterized by low soil nutrient levels (especially those of organic compounds that can provide electrons for microbial sulfate reduction1,2) and constantly oxic/hypoxic conditions.
The revegetated mine wasteland fostered new SRM genera
The occurrence of SRMs in Acidobacteria was revealed very recently14,51. To date, only 13 MAGs of SRMs in Acidobacteria (with genome completeness varying from 29.7% to 98.0%) have been reported14,15,16,51. In this context, this study not only doubled the number of such MAGs but also determined the genus-level taxonomic novelty of acidobacterial SRMs (Fig. 1). In addition, a remarkable finding of this study was that one deltaproteobacterial MAG (i.e., De.bin4) represented a genus not included among previously identified SRMs, given that SRMs in Deltaproteobacteria were investigated extensively2. It is therefore obvious that more work is needed to achieve a ‘complete understanding’ of the taxonomic diversity of SRMs, although recent studies have greatly expanded the diversity of SRMs at the OTU and phylum levels13,14. To that end, terrestrial sulfate-rich environments under constantly oxic/hypoxic conditions deserve more attention.
Acidobacterial SRMs encoded more GHs and oxygen-tolerant hydrogenases than deltaproteobacterial SRMs
Many members of Acidobacteria are thought to have the ability to use a wide range of carbohydrates, as they dedicate a large portion of their genomes to carbohydrate metabolism33. Furthermore, there was evidence that three DsrAB-encoding acidobacterial MAGs recovered from peatland sediments harbored more GH genes (on average ~105 genes per genome) than not only the other known SRMs but also the majority of non-SRM Acidobacteria (~60 genes per genome)15. Coincidently, the average number of GH genes in the six high-quality acidobacterial MAGs with reductive dsrABs in this study was up to 120 per genome (Supplementary Table 3), which was much greater than that in the three deltaproteobacterial SRMs. This result hinted at an adaptation of these acidobacterial SRMs to the oligotrophic conditions of our study site31. Notably, the most prevalent GH family across the six high-quality acidobacterial MAGs (i.e., GH3, Fig. 2) consisted of β-glucosidases (EC 3.2.1.21), β-xylosidases, and N-acetylglucosaminidases (EC 3.2.1.52)52. These enzymes can liberate glucose and xylose (i.e., growth substrates for SRMs) from plant cell wall-derived oligosaccharides (e.g., cellobiose and xylan)53. In contrast, those enzymes belonging to the most abundant GH family across the deltaproteobacterial SRMs (i.e., GH23; Fig. 2) exhibit activity towards peptidoglycan (a main component of the bacterial cell wall) and cannot release monosaccharides as a product52. Taken together, these results indicated that acidobacterial SRMs likely predominated over deltaproteobacterial SRMs in oligotrophic environments with plant residues.
Groups 1, 2a, 2b, 3a, 3b, 3c, and 3d of [NiFe]-hydrogenase as well as groups A and B1/B2 of [FeFe]-hydrogenase are known to be involved in the oxidation of H254. Two acidobacterial MAGs (i.e., Ac.bin4 and Ac.bin6, Fig. 2) lacked genes encoding such hydrogenases, suggesting their inability to couple sulfate reduction to oxidation of H21,2. This characteristic likely placed the SRMs at a disadvantage in the competition for H2 with their counterparts36 and thereby provides a possible explanation for the fact that these two MAGs occurred in our study site at a lower relative abundance than the other five high-quality MAGs possessing such genes (average 0.031% vs 0.045%; Supplementary Table 1). Remarkably, the hydrogenases encoded by the other four high-quality acidobacterial MAGs were oxygen tolerant (i.e., 1d and/or 3d of [NiFe]-hydrogenase, Fig. 2)36. This was consistent with a previous finding showing that seven acidobacterial MAGs of SRMs encoded oxygen-tolerant hydrogenases (i.e., 3b and/or 3d of [NiFe]-hydrogenase)15. Note, however, that the oxygen-sensitive D. multivorans rather than the other two oxygen-tolerant deltaproteobacterial SRMs also encoded an oxygen-tolerant hydrogenase (Fig. 2). These results suggested that the occurrence of oxygen-tolerant hydrogenases in a given SRM could improve its ability to compete for growth substrates in oxic/hypoxic environments but did not necessarily reflect its tolerance to oxygen exposure.
Acidobacterial SRMs differed from deltaproteobacterial SRMs in terms of oxygen defense mechanisms
MCPs are an indispensable component of the behavior-based antioxidant strategies (including aggregation and aerotaxis) of SRMs24,37. Among the three types of MCP genes reported previously to be involved in bacterial aerotaxis (i.e., cluster I of class Ia, class II, and class IVa)37, only class II was not recorded here. The two oxygen-tolerant deltaproteobacterial SRMs (i.e., D. vulgaris and De.bin1) harbored 17 and 18 genes encoding cluster I of class Ia MCPs, respectively, but no genes for class II and class IVa MCPs (Supplementary Table 7), indicating a predominant role of cluster I of class Ia MCPs in the aerotaxis of oxygen-tolerant deltaproteobacterial SRMs. In fact, classes II and IVa MCPs have not been previously recorded in SRMs37. Remarkably, a total of four class IVa MCPs were found in the seven oxygen-tolerant acidobacterial genomes (Supplementary Table 7). Moreover, Ac.bin5 was the only genome encoding two types of MCPs responsible for bacterial aerotaxis among the 10 focal genomes in this study (Supplementary Table 7). This result provided a possible explanation for the dominance of this acidobacterial MAG in transcripts of SRMs in the mine wasteland (Fig. 5b, d), although additional studies are needed to validate the roles of MCPs in oxygen-tolerant acidobacterial SRMs.
SOD and SOR are the main agents acting against superoxide ions in the periplasm and cytoplasm of SRMs, respectively24. The widespread distribution of genes encoding SOD across all focal SRM genomes here (Fig. 2) suggested that SOD was necessary for both deltaproteobacterial and acidobacterial SRMs to cope with oxidative stress, irrespective of whether they could tolerate oxygen in the environment. On the other hand, we found that genes encoding SOR were absent in all six high-quality MAGs of acidobacterial SRMs, despite their presence in those of deltaproteobacterial SRMs. A similar pattern was observed for TPX, an enzyme involved in peroxide detoxification in the cytoplasm of SRMs24. Therefore, our results indicated a marked difference between deltaproteobacterial and acidobacterial SRMs in terms of enzyme-based antioxidant strategies. That is, acidobacterial SRMs preferred to reduce oxygen to water before generating various types of reactive oxygen species in the cytoplasm, as they tended to have more genes encoding Cco, which can reduce oxygen to water24, but fewer genes encoding enzymes involved in detoxification of superoxide and peroxide (including SOR and TPX) than deltaproteobacterial SRMs (Fig. 2 and Supplementary Table 8).
Viral infection in SRMs was widespread
Viral infection in SRMs was first revealed by Heidelberg et al.55, who performed a whole-genome sequence analysis of D. vulgaris. This work was subsequently extended by the finding that Acidobacteria-, Candidatus Aminicenantes-, Chloroflexi-, Deltaproteobacteria-, Nitrospirae-, and Planctomycetes-related SRM genomes retrieved from wetland sediments were hosts of prophages16. Consistent with these previous studies, we revealed that five medium- to high-quality acidobacterial MAGs from the mine wasteland and the genomes of two cultured deltaproteobacterial model SRMs all contained at least one prophage (Fig. 3). Our results, along with prior findings, indicated that viral infection in SRMs was more widespread than previously thought. Remarkably, the acidobacterial SRM-specific virus/host abundance ratio recorded here approached 1:1 (Supplementary Fig. 8), which was greater than that for the phylum Acidobacteria in soils collected worldwide56. Thus, viruses infecting the acidobacterial SRMs were inferred to be in a lysogenic phase, which fitted with recent literature that suggests oligotrophic environments, such as the revegetated acidic mine wasteland, could have largely lysogenic viral populations57. On the other hand, most of the viruses recorded in this study could not be taxonomically assigned or were not closely related to any known sequenced viruses at the nucleotide level (Fig. 3), supporting the notion that the diversity of environmental viruses is largely unexplored16.
SRM-infecting viruses could contribute to glycoside hydrolysis of their hosts
Viruses are widely believed to have the ability to modulate the S biogeochemical cycle in aquatic environments, as some aquatic viruses were reported to harbor AMGs (i.e., rdsrA, rdsrC, or dsrC) encoding enzymes directly involved in dissimilatory S metabolism58,59. In contrast, little is known about the potential roles of viruses in the terrestrial S biogeochemical cycle. Here, we failed to identify viral AMGs encoding enzymes directly responsible for dissimilatory S metabolism. However, we found two viral AMGs encoding enzymes dedicated to the oxidation of organic compounds (i.e., glycoside hydrolysis; Fig. 4 and Supplementary Table 10). These results were remarkable given that no previous studies have documented such metabolic potentials of viruses hosted by SRMs and that the oxidation of organic compounds is coupled to the reduction of sulfate in SRMs1,2. In a wider context, a recent analysis of viral community composition and metabolic potentials in mangrove sediments has led to the hypothesis that viral carbohydrate AMGs may provide their hosts with energy for growth by decomposing complex carbohydrates in soil60. Interestingly, our results on the viral AMG encoding D-4,5-unsaturated β-glucuronyl hydrolase allowed us to provide a new mechanistic explanation for this hypothesis, as this enzyme can degrade plant cell wall-derived oligosaccharides to rhamnose (Fig. 4), which is accessible directly to SRMs for dissimilatory sulfate reduction47,53. With regard to endochitinase encoded by the two prophages in D. vulgaris, its products were not reported previously as substrates that could be used by SRMs for sulfate reduction (Fig. 4)53. However, some of its products (e.g., chitobiose) can be metabolized further by SRMs for use in cell wall biogenesis61. Such synergism should be advantageous for viral hosts in oligotrophic environments, shedding some light on the ‘black box’ of soil virus–host interactions57.
SRM-infecting viruses could participate in chemotaxis and antioxidation of their hosts
There was only one previous study that documented viral AMGs encoding MCPs62. We extended this work by identifying a specific host of such AMGs (i.e., D. vulgaris; Fig. 4). Moreover, our in-depth analysis of the viral MCPs recorded here revealed that they possessed an extracellular ligand-binding domain for C2/C3 carboxylates or alanine/lactate (Fig. 4 and Supplementary Table 11). Notably, most of the targeted ligands of these MCPs are growth substrates used by SRMs for sulfate reduction53. This characteristic of these MCPs could endow the host with a survival advantage in oligotrophic environments by helping the host sense its growth substrates. With regard to the viral AMG encoding the antioxidant enzyme nickel-containing superoxide dismutase (Ni-SOD; Fig. 4), none of its counterparts (i.e., the AMGs encoding various kinds of SODs) were reported previously. However, in a wider context, there were two prior reports on viral AMGs involved in antioxidation of their host. The first one showed that three AMGs (i.e., yfdK, yfdO, and yfdS) of prophages in Escherichia coli were able to enhance their host’s ability to resist oxidative stress63. The second showed that some prophages of oceanic cyanobacteria harbored genes encoding a peroxidase, which could improve the hosts’ antioxidative ability64. Thus, it is interesting to explore the generality of the involvement of viruses in chemotaxis and antioxidation of their hosts.
A new acidobacterial SRM harboring a prophage was very active
Many SRMs are known to be merely present as dormant cells under stressful environmental conditions28. To our knowledge, there was only one prior study showing clear evidence for the activity of SRMs in constantly oxic/hypoxic terrestrial environments65. In that study, SRM enumeration and geochemistry were used in combination with S isotopic analysis to infer the activity of SRMs in mine tailings. The present study provides transcriptomic evidence for the activity of SRMs (especially at the species level) in such environments. Our finding that a new acidobacterial SRM (i.e., Ac.bin5) containing a prophage dominated the transcripts of the 16 SRMs reported in this study (Fig. 5b, d) was reasonable given that, even in anoxic environments, only a minority of SRMs were metabolically active15.
We obtained several clues to understand the dominance of Ac.bin5. First, transcripts of oxygen-tolerant 1d and 3d [NiFe]-hydrogenases were detected in Ac.bin5 but not in a less active SRM (i.e., De.bin1; Supplementary Fig. 10). Second, Ac.bin5 not only encoded a complete MCP system (with two types of MCPs involved in bacterial aerotaxis) and a complete flagellum system (Fig. 2 and Supplementary Tables 6, 7) but also expressed the relevant genes (Fig. 5e), indicating that it was able to tolerate oxygen in the environment via an MCP-dependent behavior-based antioxidant strategy24. Third, Ac.bin5 harbored the greatest number of genes encoding Cco (a high-affinity oxygen reductase66) among the 10 focal genomes (Fig. 2 and Supplementary Table 8), and these genes were expressed at a level comparable to that of Cbo (another high-affinity oxygen reductase; Fig. 5e). This pattern was very different from that of D. vulgaris, in which the expression level of Cco genes under oxygen stress conditions was much lower than that of Cbo67. These findings provided further evidence for our speculation mentioned above, i.e., that acidobacterial SRMs tended to reduce oxygen to water before generating various types of reactive oxygen species in the cytoplasm. Fourth, the expression of those genes encoding enzymes responsible for scavenging superoxides and peroxides (e.g., Fe/Mn-SOD and NPX; Fig. 5e)24, along with MCP, Cco and Cbo, possibly constituted a multilayer and efficient antioxidant system for Ac.bin5. This possibility was supported by our observation that: (1) the expression level of Ac.bin5 was positively related to soil Eh but negatively related to soil Fe2+ concentration (Supplementary Fig. 9); and (2) De.bin1 was found not to transcribe genes encoding BCP, Fe/Mn-SOD and Cco (Supplementary Fig. 10). Finally, Ac.bin5 contained the second largest number of GH genes (Fig. 2 and Supplementary Table 3) and the relative expression level of GH genes was significantly higher in Ac.bin5 than in De.bin1 (Supplementary Fig. 10). These results indicated that high-level expression of GH genes could help Ac.bin5 compete for organic matter in oligotrophic environments (Supplementary Table 12)33. Despite this, the functioning of Ac.bin5 was likely inhibited by low carbon availability in the mine wasteland, as indicated by a positive relationship between the dsrAB expression level of this SRM and soil total carbon content (Supplementary Fig. 9).
Note also that the expression level of dsrAB was not significantly related to the concentrations of the main S compounds in the soil (sulfate and sulfide; P > 0.05, Supplementary Fig. 9). A similar pattern was observed for those genes responsible for the oxidation of sulfide (e.g., soxXAYZB, Supplementary Fig. 11). These results were consistent with the prior notion that interactions between S reducers and oxidizers may lead to a scenario in which end products or reaction intermediates of the S cycle remain in steady state, but this does not necessarily represent a lack of microbial S cycling17. Unfortunately, we found little transcriptomic evidence for the potential roles of SRM-infecting viruses discussed above, consistent with the result of another analysis wherein we used a newly released prophage activity estimator68 to show that the prophages recorded in this study were inactive at the sampling time point.
In conclusion, we recovered 16 reductive dsrAB-containing MAGs affiliated with Acidobacteria and Deltaproteobacteria from a revegetated sulfate-rich mine tailings pond under constantly oxic/hypoxic conditions. Among them, five were shown to represent three new SRM genera. Comparative genomics and metatranscriptomics showed that the acidobacterial and deltaproteobacterial SRMs employed different survival strategies for living in the mine wasteland. These findings supported our hypothesis. More importantly, they not only improve our understanding of the diversity and metabolic potentials of SRMs in terrestrial environments under constantly oxic/hypoxic conditions but also provide metatranscriptomic evidence for their activity in situ. Additionally, this study sheds some light on the putative roles of soil viruses in the terrestrial S biogeochemical cycle.
Methods
Study site and soil sampling
We selected a revegetated acidic mine wasteland located in southern China (29°40′52″N, 115°49′21″E) as our study site. Briefly, this site was revegetated in the spring of 2013 and consisted of three different habitats: an amended layer of revegetated tailings (0–10 cm, ALRT), an unamended layer of revegetated tailings (11–20 cm, ULRT) and unrevegetated tailings (UT). These habitats were rich in sulfate (1.80–25.9 g SO42− kg−1 dry soil) and were under constantly oxic/hypoxic conditions (as indicated by a soil Eh range of ~180–680 mV)31. Three independent soil samples for metagenomic analysis were collected from each of these habitats in July 2016 and 2017. More details on the study site, revegetation scheme, soil sampling and soil physicochemical properties are presented elsewhere31. To investigate microbial metabolic activities in situ, three additional soil samples were collected from ULRT for metatranscriptomic analysis. The soil samples were preserved in liquid nitrogen until arrival at our laboratory and were stored at −80 °C until further processing.
DNA extraction and sequence processing
The procedures used for DNA extraction, metagenomic sequencing, and data processing (including assembly, binning, refinement, genome completion estimates, gene prediction, etc.) were described in detail elsewhere31. Briefly, soil DNA was extracted using PowerSoil DNA isolation kit (Mobio Laboratories Inc., USA) with modification. The purified DNA was sequenced (250 or 150 bp paired-end reads) using an Illumina MiSeq sequencer (Illumina, USA). Raw sequencing reads were processed by eliminating duplicated reads, reads with ≥5 “N” and reads with quality score <30. The remaining high-quality reads from each metagenomics sample were assembled using SPAdes v3.9.069. The assembled scaffolds from each sample were binned using MetaBAT v0.30.3 with default parameters70. The refinement of MAGs were performed by RefineM v0.0.14 first and then by manual examination71. For the evaluation of contamination and the completeness of MAGs, CheckM v1.0.4 was used72. Gene prediction of these MAGs was conducted using Prodigal v2.6.373.
Retrieval of key genes involved in dissimilatory S metabolism
The genome-specific metabolic potential for sulfate/sulfite reduction was determined as follows. All predicted open reading frames (ORFs) in a given MAG were searched against the eggNOG74 and KEGG75 databases using Diamond76 and against HMM profiles using InterProScan77. Then, the key sulfate reduction/S oxidation genes (dsrAB, dsrD, dsrT, dsrMKJOP, aprAB, sat, and dsrEFH) in the MAGs were identified based on conserved domain hits elaborated by Anantharaman et al.14.
Phylogenetic analysis of DsrAB protein
A total of 214 DsrAB sequences (Supplementary Fig. 1), including those from both SRMs and non-SRMs reported previously14,78,79, were used for phylogenetic analysis, which could help to distinguish reductive and oxidative type DsrAB. The DsrAB sequences were aligned using MUSCLE80 with default parameters. The alignments were then filtered by TrimAL81 with the parameters -gt = 0.95 and -cons = 50. The concatenated DsrAB tree was constructed using RAxML82 with the parameters set as -f a -m PROTGAMMAIJTT –p 12345 –x 12345 -# 100. The Newick files with the best tree topology were uploaded to the Interactive Tree of Life (iTOL) online interface83 for visualization and formatting.
Sequence alignment of DsrD and DsrT proteins
The DsrD and DsrT protein sequences identified in the MAGs reported in this study were aligned along with the reference sequences respectively, using ClustalW with slow/accurate setting parameters (https://www.genome.jp/tools-bin/clustalw). The alignments were manually corrected and later visualized by ESPript 3.084. The conserved residues were highlighted.
Taxonomic classification of MAGs containing reductive dsrAB
Sixteen MAGs retrieved in our study harbored reductive dsrAB sequences (Supplementary Table 1). The direction of dissimilatory S metabolism for each MAG was determined according to the common patterns elaborated by Anantharaman et al.14. Taxonomic assignment of the 16 MAGs was inferred from the phylogenetic tree constructed with the reference genomes using GTDB-Tk85. For acidobacterial subdivision-level classification, the 12 MAGs of Acidobacteria recovered in our study were used for phylogenetic analysis with published reference genomes spanning subdivisions 1, 3, 4, 6, 8, and 2315,33. One deltaproteobacterial MAG from the Syntrophobacteraceae family without genus-level classification was used for phylogenetic tree construction with public reference genomes from Syntrophobacteraceae downloaded from GTDB-Tk. The maximum-likelihood phylogenetic trees were constructed based on a concatenated dataset of 400 universally conserved marker proteins using PhyloPhlAn86 and visualized using iTOL.
Calculation of the average AAI
The AAI values between five genomes from Syntrophobacteraceae family were calculated by AAI calculator (http://enve-omics.ce.gatech.edu/aai/index) with default parameters. The reciprocal best hits (two-way AAI) between two genomic datasets of proteins were used for further comparisons.
Calculation of relative abundances of MAGs
The relative abundances of the 16 MAGs were calculated based on the methods described elsewhere31. Briefly, the high-quality reads from each genomic dataset were mapped to all of the dereplicated MAGs using BBMap with the parameters k = 14, minid = 0.97, and build = 1. The coverage of a given MAG was calculated as the average scaffold coverage, and each scaffold was weighed by its length in base pairs. Then, the coverage of each MAG divided by the total coverage of all MAGs in each sample was considered its relative abundance.
Selection of genomes for metabolic potential analysis
Seven high-quality MAGs containing reductive dsrAB were chosen for further metabolic potential annotation, including six Acidobacteria (i.e., Ac.bin1–Ac.bin6) and one Deltaproteobacteria (i.e., De.bin1). The genome sequences of two cultured model SRMs (i.e., D. vulgaris Hildenborough (oxygen-tolerant) and D. multivorans DSM 2059 (oxygen-sensitive)35) and one cultured non-SRM species of Acidobacteria (i.e., T. gabretensis S55 isolated from oxic forest soil34), which was shown to be the species most closely related to eight out of the 12 acidobacterial MAGs in this study (Supplementary Fig. 2), were also selected for comparative genomic analysis43,55.
Identification of carbohydrate-active enzymes
To identify carbohydrate-active enzyme genes, all predicted ORFs in the 10 selected genomes were searched against the dbCAN2 meta server87 with default parameters: HMMER (E-value < 1e-15, coverage > 0.35), Diamond (evaluation < 1e-102) and Hotpep (frequency > 2.6, hits > 6). Those identified by at least two tools were kept for further classification of GH families using an in-house Perl script.
Identification of hydrogenases
For identification of hydrogenases, HMM searches were performed by searching all predicted ORFs in the 10 selected genomes. Briefly, the individual HMM profiles for [NiFe]-hydrogenases from groups 1a–1h, 2a–2d, 3a–3d, and 4a–4e, [FeFe]-hydrogenases from groups A1–A4, B and C, and Fe hydrogenase were generated using the reference sequences retrieved from a previous study36. The reference sequences were aligned using MUSCLE with default parameters, and then, the alignment was converted to Stockholm format, and databases were built using hmmscan88. The noise cut-offs for individual HMM profiles were determined by manual inspection. Protein sequences that showed the best hit with the HMM profiles with (1) bit scores greater than the calibrated threshold and (2) over 90% sequence coverage were retained.
Identification of proteins involved in respiration
All predicted ORFs in the 10 selected genomes were searched for proteins involved in four respiratory complexes based on the eggNOG annotation results (protein IDs are provided in Supplementary Table 5), including NADH dehydrogenase, succinate dehydrogenase, quinol-cytochrome-c reductase, terminal oxidase, and ATP synthase15.
Identification of proteins involved in chemotaxis and oxidative stress
In addition to MCPs, the central components of the bacterial chemotaxis system include CheA, CheB, CheR CheW, and CheY38. MCPs in the 10 selected genomes were identified by Pfam annotation hits to PF00015, while the other central protein sequences were identified by KEGG annotation hits and were further confirmed based on eggNOG annotations. Classification of MCPs was performed according to the ligand binding region and membrane topology37. Proteins involved in the core flagellum40 and type IV pilus41 systems were identified by KEGG annotation hits and were further confirmed by eggNOG annotation results. The antioxidative enzymes analyzed in this study were selected based on two previous reviews24,89 and were identified by KEGG, eggNOG and InterPro annotations.
Recovering and annotating viral scaffolds
VirSorter44 was used to recover viral scaffolds from the 16 MAGs as well as the genomes of all three cultured model species mentioned above. Only scaffolds from VirSorter categories 1, 2, 4, and 5 (categories 4 and 5 represent the provirus equivalents of categories 1 and 2) were retained. Specifically, viral scaffolds from categories 1 and 4 contain sequences similar to known viruses, and those from categories 2 and 5 contain viral hallmark genes and/or are enriched with viral or non-Caudovirales genes and have at least one other virus-like metric44,56. For scaffolds with predicted proviruses, only predicted proviral regions were retained. To taxonomically classify the viral scaffolds, a gene content-based network analysis was performed to cluster viral scaffolds into viral clusters at approximately the genus level using vConTACT2 with the ProkaryoticViralRefSeq94 database45. The ORFs in the viral scaffolds were predicted with MetaProdigal. Viral signature proteins such as terminase, integrase, capsid, and tail were identified by Pfam hits. Viral sequences that encode tail genes could be tentatively assigned to the order Caudovirales.
Estimation of viral and host abundances
The abundance of a given virus and that of its host were calculated as the normalized mean coverage depth based on the methods described elsewhere56. Briefly, the high-quality reads from each metagenomic dataset were mapped to all of the dereplicated viral scaffolds or dereplicated MAGs using BBMap with the parameters k = 14, minid = 0.97, and build = 1. The viral and host abundances were pulled from the BBMap mapping coverage output and normalized to the number of metagenomic reads in each sample.
Identification of viral AMGs
To examine the potential roles of the identified viruses in S biogeochemistry, we assessed whether they contained AMGs. The predicted viral proteins were searched against the dbCAN2 meta server87 and with HMM profiles using InterProScan77 as mentioned above. For GHs, MCPs, and Ni-SOD encoded by AMGs identified in this study, the protein sequences were structurally modeled using PHYRE290 in normal modeling mode to confirm and further resolve functional predictions.
RNA extraction and sequencing
Total cellular RNA was extracted using the RNeasy PowerSoil Total RNA kit (QIAGEN, USA) according to the manufacturer’s instructions. Total RNA was transported to the Magigene Company (Guangzhou, China) on dry ice for subsequent rRNA subtraction, cDNA synthesis, library construction, and sequencing with an Illumina NovaSeq platform (paired-end 150-bp mode).
Metatranscriptomic analysis
Raw reads were filtered by fastp91 with the parameters --cut_mean_quality 20 and -l 50. The rRNA sequences from prokaryotes and eukaryotes were removed by SortMeRNA92 with default parameters. Subsequently, the remaining reads were mapped to the genes predicted from metagenomic assemblies using BBMap with the parameters k = 14, minid = 0.97, and build = 1. The detailed information of metatranscriptomic datasets was provided in Supplementary Table 14. The expression level for each gene in each sample was normalized to reads per kilobase per million mapped reads (RPKM) values. The transcript abundance of each MAG in a given sample was calculated as the RPKM sum of all transcripts within that MAG. Similarly, the transcript abundance of a given gene from the 16 SRM-related MAGs reported in this study was calculated as the RPKM sum of all transcripts assigned to that gene from these MAGs. To compare relative gene expression levels between different SRMs, the housekeeping gene gyrA was chosen as the reference gene50. The relative expression level of the selected genes was calculated as RPKM values of the genes normalized by that of the reference gene gyrA.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The metagenomic and metatranscriptomic data analyzed in this study were deposited at EMBL under accession numbers PRJEB31441 and PRJEB42658, and the MAGs reported in this study have been deposited in GenBank under accession numbers SAMN15699825 and SAMN15808056–70.
Code availability
All software used is described in the ‘Methods’ and the code used for analysis that support findings of this study are freely available online (https://github.com/liangjl-git/SRM_scripts).
References
Barton, L. L. & Hamilton, W. A. Sulphate-reducing Bacteria: Environmental And Engineered Systems. (Cambridge University Press, Cambridge, 2007).
Rabus, R. et al. A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv. Microb. Physiol. 66, 55–321 (2015).
Rückert, C. Sulfate reduction in microorganisms — recent advances and biotechnological applications. Curr. Opin. Microbiol. 33, 140–146 (2016).
Venceslau, S. S., Stockdreher, Y., Dahl, C. & Pereira, I. A. C. The “bacterial heterodisulfide” DsrC is a key protein in dissimilatory sulfur metabolism. Biochim Biophys Acta 1837, 1148–1164 (2014).
Santos, A. A. et al. A protein trisulfide couples dissimilatory sulfate reduction to energy conservation. Science 350, 1541–1545 (2015).
Bradley, A. S., Leavitt, W. D. & Johnston, D. T. Revisiting the dissimilatory sulfate reduction pathway. Geobiology 9, 446–457 (2011).
Leavitt, W. D., Bradley, A. S., Santos, A. A., Pereira, I. A. C. & Johnston, D. T. Sulfur isotope effects of dissimilatory sulfite reductase. Front. Microbiol. https://doi.org/10.3389/fmicb.2015.01392 (2015).
Koschorreck, M. Microbial sulphate reduction at a low pH microbial sulphate reduction at a low pH. FEMS Microbiol. Ecol. 64, 329–342 (2008).
Chang, Y. J. et al. Diversity and characterization of sulfate-reducing bacteria in groundwater at a uranium mill tailings site. Appl. Environ. Microbiol. 67, 3149–3160 (2001).
Friedrich, M. W. Phylogenetic analysis reveals multiple lateral transfers of adenosine-5’-phosphosulfate reductase genes among sulfate-reducing microorganisms. J. Bacteriol. 184, 278–289 (2002).
Meyer, B. & Kuever, J. Molecular analysis of the diversity of sulfate-reducing and sulfur-oxidizing prokaryotes in the environment, using aprA as functional marker gene. Appl. Environ. Microbiol. 73, 7664–7679 (2007).
Watanabe, T., Kojima, H., Takano, Y. & Fukui, M. Diversity of sulfur-cycle prokaryotes in freshwater lake sediments investigated using aprA as the functional marker gene. Syst. Appl. Microbiol. 36, 436–443 (2013).
Vigneron, A. et al. Beyond the tip of the iceberg; a new view of the diversity of sulfite- and sulfate-reducing microorganisms. ISME J. 12, 2096–2099 (2018).
Anantharaman, K. et al. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 12, 1715–1728 (2018).
Hausmann, B. et al. Peatland Acidobacteria with a dissimilatory sulfur metabolism. ISME J. 12, 1729–1742 (2018).
Martins, P. D. et al. Viral and metabolic controls on high rates of microbial sulfur and carbon cycling in wetland ecosystems. Microbiome https://doi.org/10.1186/s40168-018-0522-4 (2018).
Bell, E. et al. Active sulfur cycling in the terrestrial deep subsurface. ISME J. 14, 1260–1272 (2020).
Colman, D. R. et al. Phylogenomic analysis of novel Diaforarchaea is consistent with sulfite but not sulfate reduction in volcanic environments on early Earth. ISME J. 14, 1316–1331 (2020).
Turaev, D. & Rattei, T. High definition for systems biology of microbial communities: metagenomics gets genome-centric and strain-resolved. Curr. Opin. Biotechnol. 39, 174–181 (2016).
Raghoebarsing, A. A. et al. A microbial consortium couples anaerobic methane oxidation to denitrification. Nature 440, 918–921 (2006).
Wrighton, K. C. et al. Metabolic interdependencies between phylogenetically novel fermenters and respiratory organisms in an unconfined aquifer. ISME J. 8, 1452–1463 (2014).
Brown, C. T. et al. Unusual biology across a group comprising more than 15% of domain Bacteria. Nature 523, 208–211 (2015).
Ljung, K., Maley, F., Cook, A. & Weinstein, P. Acid sulfate soils and human health — a millennium ecosystem assessment. Environ. Int. 35, 1234–1242 (2009).
Dolla, A., Fournier, M. & Dermoun, Z. Oxygen defense in sulfate-reducing bacteria. J. Biotechnol. 126, 87–100 (2006).
Canfield, D. E. & Des Marais, D. J. Aerobic sulfate reduction in microbial mats. Science 251, 1471–1473 (1991).
Minz, D. et al. Unexpected population distribution in a microbial mat community: sulfate-reducing bacteria localized to the highly oxic chemocline in contrast to a eukaryotic preference for anoxia. Appl. Environ. Microbiol. 65, 4659–4665 (1999).
Schoeffler, M. et al. Growth of an anaerobic sulfate-reducing bacterium sustained by oxygen respiratory energy conservation after O2-driven experimental evolution. Environ. Microbiol. 21, 360–373 (2019).
Fortin, D. & Praharaj, T. Role of microbial activity in Fe and S cycling in sub-oxic to anoxic sulfide-rich mine tailings. J. Nucl. Radiochem. Sci. 6, 39–42 (2005).
Winch, S., Mills, H. J., Kostka, J. E., Fortin, D. & Lean, D. R. S. Identification of sulfate-reducing bacteria in methylmercury-contaminated mine tailings by analysis of SSU rRNA genes. FEMS Microbiol. Ecol. 68, 94–107 (2009).
Yang, T. T. et al. Changes in microbial community composition following phytostabilization of an extremely acidic Cu mine tailings. Soil Biol. Biochem. 114, 52–58 (2017).
Liang, J. L. et al. Novel phosphate-solubilizing bacteria enhance soil phosphorus cycling following ecological restoration of land degraded by mining. ISME J. 14, 1600–1613 (2020).
Kludze, H. K., DeLaune, R. D. & Patrick, W. H. Jr. Aerenchyma formation and methane and oxygen exchange in rice. Soil Sci. Soc. Am. J. 57, 386–391 (1993).
Eichorst, S. A. et al. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ. Microbiol. 20, 1041–1063 (2018).
García-Fraile, P., Benada, O., Cajthaml, T., Baldrian, P. & Lladód, S. Terracidiphilus gabretensis gen. nov., sp. nov., an abundant and active forest soil acidobacterium important in organic matter transformation. Appl. Environ. Microbiol. 82, 560–569 (2016).
Cypionka, H., Widdel, F. & Pfennig, N. Survival of sulfate-reducing bacteria after oxygen stress, and growth in sulfate-free oxygen-sulfide gradients. FEMS Microbiol. Ecol. 1, 39–45 (1985).
Greening, C. et al. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J. 10, 761–777 (2016).
Ud-Din, A. I. M. S. & Roujeinikova, A. Methyl-accepting chemotaxis proteins: a core sensing element in prokaryotes and archaea. Cell Mol. Life Sci. 74, 3293–3303 (2017).
Wuichet, K. & Zhulin, I. B. Origins and diversification of a complex signal transduction system in prokaryotes. Sci. Signal https://doi.org/10.1126/scisignal.2000724 (2010).
Ray, J. et al. Exploring the role of CheA3 in Desulfovibrio vulgaris Hildenborough motility. Front. Microbiol. https://doi.org/10.3389/fmicb.2014.00077 (2014).
Liu, R. Y. & Ochman, H. Stepwise formation of the bacterial flagellar system. Proc. Natl Acad. Sci. USA 104, 7116–7121 (2007).
Wall, D. & Kaiser, D. Type IV pili and cell motility. Mol. Microbiol. 32, 1–10 (1999).
Crispim, J. S. et al. Screening and characterization of prophages in Desulfovibrio genomes. Sci. Rep. https://doi.org/10.1038/s41598-018-27423-z (2018).
Dörries, M., Wohlbrand, L., Kube, M., Reinhardt, R. & Rabus, R. Genome and catabolic subproteomes of the marine, nutritionally versatile, sulfate-reducing bacterium Desulfococcus multivorans DSM 2059. BMC Genomics https://doi.org/10.1186/s12864-016-3236-7 (2016).
Roux, S., Enault, F., Hurwitz, B. L. & Sullivan, M. B. VirSorter: mining viral signal from microbial genomic data. PeerJ https://doi.org/10.7717/peerj.985 (2015).
Jang, H. B. et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 37, 632–639 (2019).
Šimoliūnas, E. et al. Klebsiella phage vB_KleM-RaK2 — a giant singleton of the family Myoviridae. PLoS ONE https://doi.org/10.1371/journal.pone.0060717 (2013).
Silva, I. R., Jers, C., Meyer, A. S. & Mikkelsen, J. D. Rhamnogalacturonan I modifying enzymes: an update. N. Biotechnol. 33, 41–54 (2016).
Cohen-Kupiec, R. & Chet, I. The molecular biology of chitin digestion. Curr. Opin. Biotechnol. 9, 270–277 (1998).
Upadhyay, A. A., Fleetwood, A. D., Adebali, O., Finn, R. D. & Zhulin, I. B. Cache domains that are homologous to, but different from PAS domains comprise the largest superfamily of extracellular sensors in prokaryotes. PLoS Comput. Biol. https://doi.org/10.1371/journal.pcbi.1004862 (2016).
Zeng, L. & Burne, R. A. Sucrose-and fructose-specific effects on the transcriptome of Streptococcus mutans, as determined by RNA sequencing. Appl. Environ. Microbiol. 82, 146–156 (2016).
Flieder, M. et al. Novel taxa of Acidobacteriota implicated in seafloor sulfur cycling. ISME J. 15, 3159–3180 (2021).
Lombard, V., Ramulu, H. G., Drula, E., Coutinho, P. M. & Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 (2014).
Colleran, E., Finnegan, S. & Lens, P. Anaerobic treatment of sulphate-containing waste streams. Anton. Leeuw. Int. J. G. 67, 29–46 (1995).
Vignais, P. M. & Billoud, B. Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107, 4206–4272 (2007).
Heidelberg, J. F. et al. The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat. Biotechnol. 22, 554–559 (2004).
Emerson, J. B. et al. Host-linked soil viral ecology along a permafrost thaw gradient. Nat. Microbiol. 3, 870–880 (2018).
Parikka, K. J., Le Romancer, M., Wauters, N. & Jacquet, S. Deciphering the virus-to-prokaryote ratio (VPR): insights into virus-host relationships in a variety of ecosystems. Biol. Rev. 92, 1081–1100 (2017).
Anantharaman, K. et al. Sulfur oxidation genes in diverse deep-sea viruses. Science 344, 757–760 (2014).
Roux, S. et al. Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta- genomics. Elife https://doi.org/10.7554/eLife.03125 (2014).
Jin, M. et al. Diversities and potential biogeochemical impacts of mangrove soil viruses. Microbiome https://doi.org/10.1186/s40168-019-0675-9 (2019).
Howard, M. B., Ekborg, N. A., Weiner, R. M. & Hutcheson, S. W. Detection and characterization of chitinases and other chitin-modifying enzymes. J. Ind. Microbiol. Biotechnol. 30, 627–635 (2003).
Dinsdale, E. A. et al. Functional metagenomic profiling of nine biomes. Nature 452, 629–632 (2008).
Wang, X. X. et al. Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. https://doi.org/10.1038/ncomms1146 (2010).
Sharon, I. et al. Comparative metagenomics of microbial traits within oceanic viral communities. ISME J. 5, 1178–1190 (2011).
Praharaj, T. & Fortin, D. Indicators of microbial sulfate reduction in acidic sulfide-rich mine tailings. Geomicrobiol. J. 21, 457–467 (2004).
Morris, R. L. & Schmidt, T. M. Shallow breathing: bacterial life at low O2. Nat. Rev. Microbiol. 11, 205–212 (2013).
Lamrabet, O. et al. Oxygen reduction in the strict anaerobe Desulfovibrio vulgaris Hildenborough: characterization of two membrane-bound oxygen reductases. Microbiology 157, 2720–2732 (2011).
Kieft, K. & Anantharaman, K. Deciphering active prophages from metagenomes. mSystems https://doi.org/10.1128/msystems.00084-22 (2022).
Nurk, S., Meleshko, D., Korobeynikov, A. & Pevzner, P. A. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834 (2017).
Kang, D. D., Froula, J., Egan, R. & Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ https://doi.org/10.7717/peerj.1165 (2015).
Parks, D. H. et al. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2, 1533–1542 (2017).
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055 (2015).
Hyatt, D. et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 1–11 (2010).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Hunter, S. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 37, D211–D215 (2009).
Müller, A. L., Kjeldsen, K. U., Rattei, T., Pester, M. & Loy, A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi) sulfite reductases. ISME J. 9, 1152–1165 (2015).
Tan, S. et al. Insights into ecological role of a new deltaproteobacterial order Candidatus Acidulodesulfobacterales by metagenomics and metatranscriptomics. ISME J. 13, 2044–2057 (2019).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Capella-Gutiérrez, S., Silla-Martinez, J. M. & Gabaldon, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Letunic, I. & Bork, P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39, W475–W478 (2011).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Parks, D. H. et al. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004 (2018).
Segata, N., Börnigen, D., Morgan, X. C. & Huttenhower, C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. https://doi.org/10.1038/ncomms3304 (2013).
Yin, Y. B. et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 40, W445–W451 (2012).
Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol. https://doi.org/10.1371/journal.pcbi.1002195 (2011).
Brioukhanov, A. L. & Netrusov, A. I. Aerotolerance of strictly anaerobic microorganisms and factors of defense against oxidative stress: a review. Appl. Biochem. Microbiol. 43, 567–582 (2007).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Chen, S. F., Zhou, Y. Q., Chen, Y. R. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, 884–890 (2018).
Kopylova, E., Noe, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012).
Acknowledgements
We thank Professor AJM Baker (Universities of Melbourne and Queensland, Australia, and Sheffield, UK) for his help in the improvement of this paper. This work was supported financially by the National Natural Science Foundation of China (Nos. 42177009, 41622106, 42077117, and 41603074), the Natural Science Foundation of Guangdong Province of China (Nos. 2020A1515010937 and 2020A1515110972), and the Key-Area Research and Development Program of Guangdong Province (No. 2019B110207001).
Author information
Authors and Affiliations
Contributions
J.T.L. and J.L.L. designed all the experiments and wrote the manuscript. P.J., X.J.W., S.NOu, T.T.Y., S.W.F., J.L.L., Z.F., and J.L. were responsible for performing the field and laboratory experiments. J.T.L., P.J., B.L., W.S.S., and J.L.L. analyzed all the data, discussed the results and critically reviewed the manuscript. All authors read the final manuscript and approved its publication.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, Jt., Jia, P., Wang, Xj. et al. Metagenomic and metatranscriptomic insights into sulfate-reducing bacteria in a revegetated acidic mine wasteland. npj Biofilms Microbiomes 8, 71 (2022). https://doi.org/10.1038/s41522-022-00333-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41522-022-00333-9
This article is cited by
-
Hidden diversity and potential ecological function of phosphorus acquisition genes in widespread terrestrial bacteriophages
Nature Communications (2024)
-
Oxygen respiration and polysaccharide degradation by a sulfate-reducing acidobacterium
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
