
Abstract
Creatine transporter (CT) deficiency is an X-linked disorder caused by mutations in the SLC6A8 gene. We describe a clinical, biochemical and molecular examination of a child with X-linked cerebral creatine deficiency. Increased urinary creatine/creatinine ratio, abnormal brain proton magnetic resonance spectroscopy and reduced creatine transport confirmed the clinical diagnosis. SLC6A8 analysis revealed a novel mutation that was hemizygous in the child and not detected in his mother. CT deficiency should be considered in children, especially males, with mental retardation.
Similar content being viewed by others


Creatine transporter (CT) deficiency (OMIM 300036) has been reported to be the most common cerebral creatine deficiency syndrome (CCDS) and may be one of the leading causes of X-linked mental retardation (X-LMR) in males, with a prevalence of 2.1% according to a European X-LMR panel.1,2 Clinical symptoms include delays in speech and language development, intellectual disabilities, autistic-like behavior and in some cases, seizures. However, clinical severity greatly varies, with some children exhibiting marked developmental delay and seizures and others having more modest clinical involvement.3 X-LMR is caused by mutations in the SLC6A8 gene, a member of the solute-carrier family 6 mapped to Xq28.4 The transporter encoded by this gene, CT1, is a multi-pass membrane protein required for the uptake of creatinine in muscles and the brain. CT1 is widely expressed in brain tissue, predominantly in the cortical and subcortical regions involved in motor and sensory processing, learning, memory, and regulation of emotion-related behavior.5 To date, numerous mutations in the SLC6A8 gene have been described.6,7
The diagnosis of brain creatine deficiency is based on clinical presentation, an increased urinary creatine/creatinine (Cr/Crn) ratio, and abnormal brain Cr content assessed by brain magnetic resonance spectroscopy (MRS).8 Nevertheless, brain MRS is not always feasible, and diagnosis is commonly based on clinical presentation and/or the presence of biochemical markers, such as an increased urinary Cr/Crn ratio and differential diagnosis for CT deficiency from other CCDS. The diagnosis is confirmed by measuring long-term Cr accumulation in fibroblasts or via DNA testing.7 Here we describe a case of a boy who initially exhibited motor dysfunction and hypotonia and who presented with a new splicing mutation in the SLC6A8 gene.
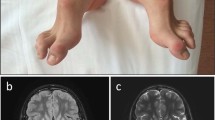
The patient was a 12-year-old Caucasian male born at term after an uneventful pregnancy, with an Apgar score of 9–10 and normal weight, length, and head circumference at birth. The parents were both unrelated and healthy. The patient was referred to the neuropediatric unit for evaluation and early intervention for mild hypotonia and motor delay at 15 months of age. He walked independently and uttered the first words at the age of 3 years. Simple focal seizures with secondary generalization were present from 2 to 6 years of age, and most seizures were of short duration, febrile, and non-febrile and well controlled with antiepileptic drugs. At 6 years old, the patient was submitted to a comprehensive neuropsychological evaluation to assess cognitive, speech, and social and personal ability. The Battelle Developmental Inventory total developmental quotient score was 60, with discrepancy between nonverbal and verbal skills; he exhibited qualitative communication difficulties on the Autism Diagnostic Interview-Revised. Video electroencephalography recording revealed multifocal spike and polispike waves at right frontal, temporal, and posterior regions. As then, changes in the frequency and complexity of seizures, abnormal visual sensation (colored lights), and abdominal pain and nausea have been observed. Currently, he exhibits moderately delayed motor development and learning, slight hyperactivity, and much better behavior. Seizures occur every 3–4 months.
Biochemical screening was performed with blood and urine samples. The Cr/Crn ratio was determined in two urine samples taken on different days. Cr uptake was assayed in fibroblasts after incubation for 24 h in medium containing physiological and supraphysiological Cr concentrations (25 and 500 μM, respectively) according to a previously reported procedure.9 An abnormal Cr/Crn ratio was observed in the two urine samples (2.66 and 4.56: reference value <1.5). Cr uptake in fibroblasts was nearly undetectable in culture when incubated at a physiological Cr concentration. Cr uptake after incubation with supraphysiological levels of Cr was <25% compared with control cells, indicating impaired Cr uptake in fibroblasts.
Brain magnetic resonance imaging (MRI) and proton MRS (1H-MRS) were performed to confirm the diagnosis. MRI showed hyperintense T2/FLAIR signals at the periventricular bifrontal white matter and semioval centers, and 1H-MRS revealed a marked reduction of Cr peak.
Molecular analysis of blood samples from the proband and his parents were obtained after obtaining informed consent. Genomic DNA and RNA were obtained from peripheral blood using QIAamp DNA Mini Kit and QIAamp RNA Blood Mini Kit, respectively (QIAGEN GmgH, Hilden, Germany). Karyotype, CGH array 60 K (Agilent Technologies, California, USA) and a molecular study of SCN1A and FMR1 genes were performed as part of clinical care, and all were normal. Multiplex ligation-dependent probe amplification (MLPA) (SALSA MLPA kit P049-B2, MRC-Holland, Amsterdam, Netherlands) PCR and reverse transcription–PCR, including direct gene sequencing of all exons and the flanking intronic sequences of the SLC6A8 gene, were conducted. Regarding the molecular analysis, MLPA showed an aberrant pattern. DNA and cDNA sequence analysis of the SLC6A8 gene revealed a 34-base pair (bp) deletion (c.1016+11_1017-52del, intron 6) at the DNA level that produced a 30-bp deletion in exon 7 (r.1017_1046del) in cDNA (Figure 1). The variant was considered pathogenic by splice-site analysis with the NetGene2 Server (http://www.cbs.dtu.dk/services/NetGene2/) and Human Splicing Finder (http://www.umd.be/HSF/). The variant was not found in 100 control samples. The mutation was not detected in the mother, and therefore, it was considered de novo. However, somatic mosaicism cannot be excluded; family counseling and prenatal diagnosis during further pregnancies should be offered. This splicing mutation variant has not been previously described, and splicing mutations in SLC6A8 are less common, as reported by van de Kamp et al.7

Schematic representation of direct DNA (a) and cDNA (b) sequencing. (a) shows the DNA sequence of the patient and his mother. The blue arrow indicates the exact point of the 34-base pair (bp) deletion (c.1016+11_1017-52del, intron 6). (b) shows the cDNA sequence of the patient and his mother and the blue arrows highlight the 30-bp deletion in exon 7 (r.1017_1046del).
Disorders of creatine synthesis and cellular transport result in brain creatine deficiency and represent a relatively novel cause of mental retardation and seizures. Patients with X-linked CT deficiency can have different clinical presentations, even within the same family, with developmental delays/mental retardation being present in all patients. This disease is suspected to be due to reduced brain creatine content, as detected by MRS. The increased Cr/Crn ratio in urine further verifies the diagnosis.
Treatment of patients with CT deficiency remains ineffective. In addition, cyclocreatine treatment in brain-specific SCL6A8 knockout mice, an animal model of human CT deficiency, showed promising results.10 The identification of new cases of CT deficiency is important, and the inclusion of Cr/Crn urine analysis in screening patients with these neurological symptoms should be considered.11 Rosenberg et al.12 recommended that once increased Cr/Crn is detected, the diagnosis of CT deficiency should be confirmed by functional studies and/or DNA testing.
In conclusion, we report a case of a moderate form of CT deficiency caused by a novel SLC6A8 mutation. Information about genotype–phenotype correlations is important for understanding genetic disorders such as CT deficiency.
References
References
Clark AJ, Rosenberg EH, Almeida LS, Wood TC, Jakobs C, Stevenson RE et al. X-linked creatine transporter (SLC6A8) mutations in about 1% of males with mental retardation of unknown etiology. Hum Genet 2006; 119: 604–610.
Rosenberg EH, Almeida LS, Kleefstra T, deGrauw RS, Yntema HG, Bahi N et al. High prevalence of SLC6A8 deficiency in X-linked mental retardation. Am J Hum Genet 2004; 75: 97–105.
Stevenson RE, Schwartz CE, Schroer RJ . X-linked Mental Retardation. Oxford University Press: New York, 2000.
Salomons GS, van Dooren SJ, Verhoeven NM, Cecil KM, Ball WS, Degrauw TJ et al. X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am J Hum Genet 2001; 68: 1497–1500.
Cecil KM, Salomons GS, Ball WSJ, Wong B, Chuck G, Verhoeven NM et al. Irreversible brain creatine deficiency with elevated serum and urine creatine: a creatine transporter defect? Ann Neurol 2001; 49: 401–404.
Betsalel OT, Rosenberg EH, Almeida LS, Kleefstra T, Schwartz CE, Valayannopoulos V et al. Characterization of novel SLC6A8 variants with the use of splice-site analysis tools and implementation of a newly developed LOVD database. Eur J Hum Genet 2011; 19: 56–63.
van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S, Abulhoul L, Grünewald S, Anselm I et al. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J Med Genet 2013; 50: 463–472.
Dreha-Kulaczewski S, Kalscheuer V, Tzschach A, Hu H, Helms G, Brockmann K et al. A novel SLC6A8 mutation in a large family with X-linked intellectual disability: clinical and proton magnetic resonance spectroscopy data of both hemizygous males and heterozygous females. JIMD Rep 2014; 13: 91–99.
Arias A, Corbella M, Fons C, Sempere A, García-Villoria J, Ormazabal A et al. Creatine transporter deficiency: prevalence among patients with mental retardation and pitfalls in metabolite screening. Clin Biochem 2007; 40: 1328–1331.
Stockler S, Schutz PW, Salomons GS . Cerebral creatine deficiency syndromes: clinical aspects, treatment and pathophysiology. Subcell Biochem 2007; 46: 149–166.
Almeida LS, Verhoeven NM, Roos B . Creatine and guanidinoacetate: diagnostic markers for inborn errors in creatine biosynthesis and transport. Mol Genet Metab 2004; 82: 214–219.
Rosenberg EH, Martinez Munoz C, Betsalel OT, van Dooren SJ, Fernandez M, Jakobs C et al. Functional characterization of missense variants in the creatine transporter gene (SLC6A8): improved diagnostic application. Hum Mutat 2007; 28: 890–896.
Data Citations
Cristina, Cervera-Acedo HGV Database (2015) http://dx.doi.org/10.6084/m9.figshare.hgv.702
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Cervera-Acedo, C., Lopez, M., Aguirre-Lamban, J. et al. A novel SLC6A8 mutation associated with motor dysfunction in a child exhibiting creatine transporter deficiency. Hum Genome Var 2, 15037 (2015). https://doi.org/10.1038/hgv.2015.37
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/hgv.2015.37
