
Abstract
Thiamine metabolism dysfunction syndrome-4 (THMD4) includes episodic encephalopathy, often associated with a febrile illness, causing transient neurologic dysfunction and a slowly progressive axonal polyneuropathy. Until now only two mutations (G125S and S194P) have been reported in the SLC25A19 gene as causative for this disease and a third mutation (G177A) as related to the Amish lethal microcephaly. In this work, we describe the clinical and molecular features of a patient carrying a novel mutation (c.576G>C; Q192H) on SLC25A19 gene. Functional studies on this mutation were performed explaining the pathogenetic role of c.576G>C in affecting the translational efficiency and/or stability of hMTPPT protein instead of the mRNA expression. These findings support the pathogenetic role of Q192H (c.576G>C) mutation on SLC25A19 gene. Moreover, despite in other patients the thiamine supplementation leaded to a substantial improvement of peripheral neuropathy, our patient did not show a clinical improvement.
Similar content being viewed by others

Introduction
Thiamine (vitamin B1) is an essential micronutrient that, in its biologically active forms (thiamine pyrophosphate, TPP), acts as a cofactor for critical metabolic reactions related to carbohydrate and energy metabolism. Thiamine also plays an important role in reducing cellular oxidative stress via maintaining normal cellular redox state. Low cellular levels of thiamine lead to an impairment in oxidative energy metabolism (acute energy failure), oxidative stress, and in the structure/function of mitochondria [1]. Thiamine requires specific transporter for the absorption in intestine (thiamine transporter-1 and thiamine transporter-2) and mitochondrial uptake (mitochondrial TPP transporter (hMTPPT)), encoded by SLC25A19). In recent years, genetic defects in thiamine transport and metabolism has been reported [2]. Among them, thiamine metabolism dysfunction syndrome-4 (THMD4) (OMIM #613710) represents an autosomal recessive ultrarare metabolic disorder characterized by childhood onset of episodic encephalopathy, often associated with a febrile illness, and causing transient neurologic dysfunction. Most of patients fully recover, but someone may have mild residual weakness. Moreover during childhood, patients develop a slowly progressive axonal polyneuropathy. Brain imaging during the acute episodes shows lesions consistent with bilateral striatal degeneration or necrosis [2, 3].
The disease is caused by mutations in the mitochondrial TPP uptake carrier SLC25A19 gene. Until now, only two mutations (G125S and S194P) have been reported in homozygous state in this gene. A third mutation (G177A) found in SLC25A19 accounts for the Amish lethal microcephaly (MCPHA; OMIM #607196), a disorder with a more severe phenotype [4].
In this report, we describe the clinical and molecular features of a patient carrying a novel mutation (c.576G>C; Q192H) in SLC25A19 also providing functional studies supporting its pathogenicity.
Clinical description
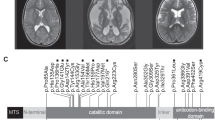
The boy is the fourth child of a consanguineous Italian mating. The maternal grandfather of the boy’s mother was the brother of the paternal grandmother of the father (fifth degree of consanguinity) (Supplemental Material). The boy showed a history of neurological impairment due to an encephalopathy during fever. He had four acute events started during a fever episode when he was 1 year old, and characterized by tremors, hypotonia, gait difficulty, dysphagia, dysphonia, and alteration of consciousness. Now he is 18 years old and he presents a moderate cognitive delay, bilateral pes cavus with hammer toes (Fig. 1a), and atrophy of intraosseous muscles. Brain MRI shows multiple focal areas of signal alteration with caudate nucleus, putamen and cerebellum involvement, and slight symmetrical atrophy of the insula cortex (Fig. 1b, c). Electromyography showed a motor and sensory neuropathy, while ECG was normal. After the diagnosis at 18 years of age the proband started a treatment with thiamine supplementation at the dose of 400 mg/day without relevant clinical improvement after 1 year of follow-up in terms of peripheral neuropathy, gait difficulty, and cognitive delay.

Clinical features. a Bilateral pes cavus with hammer toes. b MRI brain showing multiple focal areas of signal alteration and slight symmetrical atrophy of the insula cortex
Materials and methods
Ethical approval and informed consent
All experimental protocols were approved by the ethical committee of IRCCS “Burlo Garofolo” hospital. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent was obtained from all individual participants and/or their legal guardian/s included in the study.
SNP-array analysis and mutation screening
Homozygosity mapping was performed using HumanCytoSNP-12 DNA Analysis BeadChip (Illumina, CA). Data obtained were analyzed with Plink [5] software using the following parameters: length of the sliding window 1/41,000 kb; number of SNPs in a window 1/4100 and a minimum density of 1 SNP/kb. Specific primers for amplification of the coding region and exon–intron boundaries of the SLC25A19 gene were designed (sequences available upon request). Direct sequencing of the PCR products were performed by standard procedures using an ABI PRISM 3110 Genetic Analyzer (Life Technologies, CA). All variants were checked on the Exac, dbSNP, and HGMD professional databases. Pathogenicity of missense mutation were predicted with several online pathogenicity prediction tools as in Bottega et al. [6].
Comparative protein modeling and computational analysis
The hMTPPT polypeptide sequence was subjected to Phyre2 fold recognition program (http://www.sbg.bio.ic.ac.uk/phyre2) obtaining an homology model that was selected according to criteria previously described [7]. That model was designed based on “mitochondrial ADP/ATP carrier” (PDB ID: 1OKC) as the most suitable template [8]. The template gives 93% coverage for hMTPPT with 100% confidence. The generated three-dimensional (3D) structure of hMTPPT was visualized by PyMOL (https://www.pymol.org/). The topology of hMTPPT determined from its secondary structure and predicted by “TMpred” program (http://www.ch.embnet.org/software/TMPRED_form.html) matches the relative orientation of amino acids as determined by comparative 3D modeling. Conserved amino acids residues were identified by multiple sequence alignment using “PRALINE” software (www.ibi.vu.nl/programs/pralinewww/) [9].
Site-directed mutagenesis and overexpression of hMTPPT wild-type (WT) and clinical mutant
The mutation c.576G>C was introduced by site-direct mutagenesis in the open reading frame of hMTPPT (SLC25A19 gene; in a pEGFP plasmid) using the Quick change™ II site-directed mutagenesis kit (Santa Clara, CA) and specific primers (Forward: 5′-TACGCCGGGCTGCACTTCTCTTGCTACAG-3′; Reverse: 5′-CTGTAGCAAGAGAAGTGCAGCCCGGCGTA-3′) as described previously [10]. The mutated construct was confirmed by DNA sequencing (Laragen, Los Angeles, CA).
For stable selection of WT and Q192H hMTPPT plasmids, HepG2 cells (ATCC, Manassas, VA) at 80–90% confluence were transiently transfected with 10 µg of WT or the mutant (Q192H) hMTPPT using 10 µl of Lipofectamine2000 (Invitrogen, CA). Then, cells were selected using G418 (0.5 mg/ml; Invitrogen) for 6–8 weeks as described previously [11]; overexpression of both of the hMTPPT proteins was confirmed by western blotting.
Isolation of mitochondria and uptake measurement
Mitochondria were isolated from HepG2 cells stably expressing WT hMTPPT and Q192H hMTPPT constructs using mitochondria isolation kit (Thermo Scientific, Rockford, IL). Uptake of 3H-thiaminpyrophosphate (TPP, Moravek Biochemicals, Brea, CA) by freshly isolated mitochondria was performed as described previously [7, 11]. Protein concentration of the mitochondrial suspensions was determined by DC protein assay kit (Bio-Rad).
Western blot analysis
WT and Q192H mutated hMTPPT proteins were identified by western blot analysis as described previously [12]. Anti-GFP (Clontech, Montain view, CA) and anti-pyruvate dehydrogenase (PHD, Abcam, Cambridge, MA) monoclonal antibodies were used. Visualization of the bands was performed using an Odyssey infrared imaging system (LI-COR Bioscience). Odyssey application software (version 3.0) was used to determine density of the individual band.
Real-time PCR
Total RNA was isolated from HepG2 cells stably expressing the WT and the mutant hMTPPT using TRIzol, treated with DNase I, and subjected to reverse transcription (iScript cDNA synthesis kit; Bio-Rad, Hercules, CA). Level of mRNA was quantified in a CFX96 real-time PCR system (Bio-Rad), using iQ SYBR Green Super mix (Bio-Rad) and specific hMTPPT primers as described previously [12]. Data were normalized relative to the internal control B-actin and calculated using a relative relationship method [13].
Statistical analysis
Data are mean ± SE of at least three separate determinations. Uptake was expressed in terms of fmol/mg protein/5 min. Statistical analysis was done using the Student’s t test; significance was set at P < 0.05.
Results
Consanguinity evaluation and mutational screening
SNPs-array analysis was performed in our patient to study the presence of runs of homozygosity (ROHs) since an inbreeding mating was reported. Consistently, SNPs-array analysis revealed many areas of homozygosity, as in consanguinity. Eighteen ROHs turned out to be larger than 1 Mb. The SLC25A19 gene was within a 5.7 Mb ROH on chromosome 17 (chr17:71962659-77748961). The subsequent Sanger sequencing of SLC25A19 gene showed the homozygous mutation c.576G>C (Q192H) in exon 6. Segregation analysis, confirmed the presence of the same mutation in heterozygous state in both parents. Variant is not reported in pathogenicity (HGMD professional) and healthy control databases (ExAC, 1000G, GnomAD). It was predicted to be likely pathogenic according to the ACMG/AMP guidelines and by all the pathogenicity prediction tools used (MutationTaster, score = 0.999, “disease causing”; PolyPhen-2, score = 0.880, “possibly damaging” and CADD, score = 27.1, “damaging”).
Localization of the Q192H mutation and predicted effect on protein structure
The hMTPPT (SLC25A19) protein consists in 320 amino acids (~35 kDa) organized into six transmembrane domains (TMDs) with both the N- and C-termini phasing the cytoplasm [7]. Analysis of the hMTPPT polypeptide membrane topology prediction locate the newly identified mutation (Q192H) at the fourth protein TMD (Fig. 2a). Interestingly, position 192 of the hMTPPT protein (i.e., the glutamine) appeared to be conserved among species (Fig. 2b).

Localization of the Q192H mutation and predicted effect on protein structure. a Predicted membrane topology of hMTPPT protein and location of the Q192H clinical mutation. The hMTPPT is predicted to have six potential TMDs. Arrows indicate the location of clinical mutation Q192H. b Conservativeness of the amino acid at position 192 of the hMTPPT among different species. The polypeptide sequence of MTPPT from human (NP_001119594), pongo (NP_001127123), bovine (NP_001039352), mouse (NP_001239313), rat (NP_001007675), and yeast (NP_011610) were aligned using PRALINE program. Conserved and unconserved residues are indicated by red to blue colors, respectively; location of the clinical mutation (in the fourth TMD at position 192) is shown by dotted box. c Structural comparison of the Q192H clinical mutant and the WT hMTPPT. The 3D structure of hMTPPT (green color) and its clinical mutant (pink color) was generated based on the PDB ID entry 1OKC (i.e., a crystal structure of the mitochondrial ADP/ATP carrier in complex with carboxyatractyloside) and superimposed both the 3D structure. The structural changes are indicated by black arrow
In order to determine if the newly identified Q192H mutation leads to change(s) in the protein tertiary structure, we generated a 3D homology model of the WT and the Q192H mutated hMTPPT using Phyre2 server analysis (see section “Materials and methods”) and superimposed the two models. The result showed that the mutation leads to a substantial change in the hMTPPT tertiary structure folding (Fig. 2c).
Effect of the Q192H mutation on protein functionality
In order to examine the effect of identified mutation on protein functionality, a pEGFP expression vector containing the WT hMTPPT (wtMTPPT) protein was mutagenized in order to express the mutated (Q192H) form of the protein. HepG2 cells stably expressing the WT and mutated hMTPPT constructs were seeded for mitochondria isolation to be tested for 3H-TPP (0.38 μM) uptake evaluation. The results showed a significant (p < 0.01) decreasing in 3H-TPP uptake by mitochondria from cells expressing the mutant compared to the WT form of hMTPPT (Fig. 3a).

Effect of newly identified clinical mutation (Q192H) in the hMTPPT on TPP uptake. a Uptake of 3H-TPP (0.38 μM; 5 min; pH 7.4) by freshly isolated mitochondria from HepG2 cells stably expressing the WT hMTPPT and the clinical mutant Q192H. Data are mean ± SE of at least three independent experiments, **P < 0.01. Effect of the Q192H mutation on level of expression of hMTPPT mRNA (b) and protein (c, d). b mRNA levels were determined by real-time PCR. Data are mean ± SE of three to four independent experiments (normalized relative to β-actin). *P < 0.05. c, d show western blot analysis performed using identical amount of total HepG2 cell homogenate and freshly isolated mitochondrial suspension, respectively. Plots were probed with anti-GFP monoclonal antibody and with anti-PDH monoclonal antibody; data were normalized relative to the mitochondrial internal control, PDH. Representative western blots are shown in inset. Data are mean ± SE of at least three different samples from three different batches of cells. **P < 0.01
To determine whether the reduction in TPP uptake by the mutant hMTPPT is due to a decrease in mRNA expression, we performed a RT-PCR assay on RNA obtained from HepG2 cells. Comparison of the mRNA expression level from cells expressing the WT or the mutated hMTPPT form reveals no significative differences in mRNA level (Fig. 3b).
As the mutation does not affect mRNA expression level, we investigated whether the Q192H mutation leads to translation efficiency/protein stability impairment. A western blot analysis was performed using whole cell homogenate from HepG2 cells. The results showed that the Q192H protein level is significantly (p < 0.01) lower than that of the WT hMTPPT (Fig. 3c). Collectively, these findings suggest that while the Q192H mutation has no effect on expression of the hMTPPT mRNA, it does affect translational efficiency and/or stability of the hMTPPT protein.
Then, we examined the expression of the mutant hMTPPT protein in mitochondria in order to investigate whether protein targeting/delivery is impaired due to Q192H mutation. Equal amounts of purified mitochondria (∼60 μg) from HepG2 cells stably expressing the mutant and the WT hMTPPT were analyzed by western blot. The results showed a significantly (p < 0.01) decrease of mutated protein expression in mitochondria compared to the WT protein (Fig. 3d).
Discussion
The proband showed several episodes of encephalopathy with hypotonia and progressive polyneuropathy. The clinical picture, jointly with MRI images and the presence of consanguinity, confirmed by ROH analyses, allowed to associate the SLC25A19 mutation with the clinical features of the boy. The SLC25A19 gene encodes the 320-residue mitochondrial TPP transporter (hMTPPT) [14]. TPP, the activated form of thiamine, is an essential cofactor of three thiamine-dependent mitochondrial enzymes (PHD, branched chain 2-oxo acid dehydrogenase and the oxoglutarate dehydrogenase complexes that catalyze essential reactions involving the oxidative energy metabolism [15]. Mutations in hMTPPT carrier lead to a drastic depletion in mitochondrial TPP level, which is causative for two different clinical diseases: the Amish microcephaly, reported for the only c.530G>C (p.G177A) mutation [4] and the THMD4, accounting until now for only two reported mutations (c.373G>A, G125S and c.580T>C, S194P) [2, 3]. The milder symptoms reported for THMD4 in comparison to the Amish microcephaly might suggest that the some aminoacidic changes are better tolerated in hMTPPT structure.
The other five reported patients with THMD4 belong from two unrelated families (three sisters and a brother for one family [3] and only the proband from the second family [2]). In both cases parents were consanguineous and the causative mutations (c.373G>A and c.508T>C respectively) were found in homozygous state. Heterozygous parents and a reported sister from the first family were healthy. All patients share similar clinical features as the onset of clinical signs during infancy (20 months–6 years) when each of them suffered from episodes of acute weakness culminating in flaccid paralysis and encephalopathy. Concomitant with the weakness and areflexia, obtundation, aphasia, and disorientation became evident. Laboratory tests revealed elevated levels of lactate acid in the cerebrospinal fluid during the acute phase but normal systemic mitochondrial biomarkers [2, 3]. Brain MRI revealed T2 hyperintense lesions and necrosis distributed in the putamen and the caudate nuclei. Axonal damage, more pronounced in the lower limbs, with decreased amplitude on electrophysiologic studies [2, 3]. Our proband shows clinical overlapping features and MRI images comparing to the other reported THMD4 patients, suggesting a low clinical and phenotypic variability of these patients.
Our functional analysis on hMTPPT explain the pathogenesis of this mutation suggesting that while the Q192H mutation of SLC25A19 has no effect on mRNA expression, it does affect the translational efficiency and/or stability of the hMTPPT protein.
Finally, although in patients described by Ortigoza-Escobar et al. [2], the thiamine supplementation leads to substantial improvement of peripheral neuropathy allowing a milder phenotype in treated patients, our patient did not show the expected outcome. Of course, since the parents of our patient are consanguineous, we can not exclude a deleterious effect of other homozygous variants in his genome.
In conclusion, we describe in this report the third mutation (c.576G>C; Q192H) of the SLC25A19 gene and demonstrate its impact on hMTPPT function broadening the knowledge on the THMD4 patients and its pathogenesis.
References
Bettendorff L, Goessens G, Sluse F, Wins P, Bureau M, Laschet J, et al. Thiamine deficiency in cultured neuroblastoma cells: effect on mitochondrial function and peripheral benzodiazepine receptors. J Neurochem. 1995;64:2013–21.
Ortigoza-Escobar JD, Alfadhel M, Molero-Luis M, Darin N, Spiegel R, de Coo IF, et al. Thiamine deficiency in childhood with attention to genetic causes: survival and outcome predictors. Ann Neurol. 2017;82:317–30.
Spiegel R, Shaag A, Edvardson S, Mandel H, Stepensky P, Shalev SA, et al. SLC25A19 mutation as a cause of neuropathy and bilateral striatal necrosis. Ann Neurol. 2009;66:419–24.
Rosenberg MJ, Agarwala R, Bouffard G, Davis J, Fiermonte G, Hilliard MS, et al. Mutant deoxynucleotide carrier is associated with congenital microcephaly. Nat Genet. 2002;32:175–9.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Bottega R, Nicchia E, Cappelli E, Ravera S, De Rocco D, Faleschini M, et al. Hypomorphic FANCA mutations correlate with mild mitochondrial and clinical phenotype in Fanconi anemia. Haematologica. 2018;103:417–26.
Sabui S, Subramanian VS, Kapadia R, Said HM. Structure-function characterization of the human mitochondrial thiamin pyrophosphate transporter (hMTPPT; SLC25A19): important roles for Ile(33), Ser(34), Asp(37), His(137) and Lys(291). Biochim Biophys Acta. 2016;1858:1883–90.
Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trézéguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature. 2003;426:39–44.
Bawono P, Heringa J. PRALINE: a versatile multiple sequence alignment toolkit. Methods Mol Biol. 2014;1079:245–62.
Ravera S, Dufour C, Cesaro S, Bottega R, Faleschini M, Cuccarolo P, et al. Evaluation of energy metabolism and calcium homeostasis in cells affected by Shwachman-Diamond syndrome. Sci Rep. 2016;6:25441.
Subramanian VS, Nabokina SM, Lin-Moshier Y, Marchant JS, Said HM. Mitochondrial uptake of thiamin pyrophosphate: physiological and cell biological aspects. PLoS ONE. 2013;8:e73503.
Subramanian VS, Subramanya SB, Said HM. Relative contribution of THTR-1 and THTR-2 in thiamin uptake by pancreatic acinar cells: studies utilizing Slc19a2 and Slc19a3 knockout mouse models. Am J Physiol Gastrointest Liver Physiol. 2012;302:G572–8.
De Rocco D, Melazzini F, Marconi C, Pecci A, Bottega R, Gnan C, et al. Mutations of RUNX1 in families with inherited thrombocytopenia. Am J Hematol. 2017;92:E86–8.
Lindhurst MJ, Fiermonte G, Song S, Struys E, De Leonardis F, Schwartzberg PL, et al. Knockout of Slc25a19 causes mitochondrial thiamine pyrophosphate depletion, embryonic lethality, CNS malformations, and anemia. Proc Natl Acad Sci USA. 2006;103:15927–32.
Foulon V, Antonenkov VD, Croes K, Waelkens E, Mannaerts GP, Van Veldhoven PP, et al. Purification, molecular cloning, and expression of 2-hydroxyphytanoyl-CoA lyase, a peroxisomal thiamine pyrophosphate-dependent enzyme that catalyzes the carbon-carbon bond cleavage during alpha-oxidation of 3-methyl-branched fatty acids. Proc Natl Acad Sci USA. 2009;96:10039–44.
Acknowledgements
This study was supported by IRCCS Burlo Garofolo (Ricerca Corrente 31/17). The authors thank the family for their participation in this project.
Author information
Authors and Affiliations
Contributions
RB, MDP, HMS, and FF designed the study, and drafted the manuscript. SS and RB performed functional studies under HMS and FF supervision. VP performed SNP-array analysis. MDP, KV, AT, and FF collected the samples and performed clinical analysis. All authors revised critically the paper and approved the final version.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Bottega, R., Perrone, M.D., Vecchiato, K. et al. Functional analysis of the third identified SLC25A19 mutation causative for the thiamine metabolism dysfunction syndrome 4. J Hum Genet 64, 1075–1081 (2019). https://doi.org/10.1038/s10038-019-0666-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s10038-019-0666-5
This article is cited by
-
Identification and functional analysis of novel SLC25A19 variants causing thiamine metabolism dysfunction syndrome 4
Orphanet Journal of Rare Diseases (2021)
