
Abstract
Extant patterns of population structure and levels of diversity are a consequence of factors that vary in both space and time. Our objective in this study is to investigate a species that has responded to both natural and anthropogenic changes in ways that have shaped modern populations and provide insight into the key processes. The roe deer (Capreolus capreolus) is one of the two species of deer native to Britain. During the last glacial maximum (LGM), the British habitat was largely under ice and there was a land bridge to mainland Europe. As the Earth warmed during the early Holocene, the land bridge was lost. Subsequent hunting on the British mainland left the southern region extirpated of roe deer, whereas a refugial population remained in the north. Later reintroductions from Europe led to population expansion, especially in southern United Kingdom. Here, we combine data from ancient and modern DNA to track population dynamics and patterns of connectivity, and test hypotheses about the influence of natural and anthropogenic environmental change. We find that past expansion and divergence events coincided with a warming environment and the subsequent closure of the land bridge between Europe and the United Kingdom. We also find turnover in British roe deer haplotypes between the late-Holocene and modern day that have likely resulted from recent human disturbance activities such as habitat perturbation, overhunting and restocking.
Similar content being viewed by others
Introduction
Evolutionary process determines the level and structure of genetic diversity within and between natural populations. This is influenced by both spatial and temporal factors, and both adaptation and genetic drift. Understanding the relationship between specific pressures on populations and the consequences for evolutionary potential is a core objective of conservation genetics, as well as an essential aspect of understanding evolution. In this study we focus on how environmental factors (including those of anthropogenic origin) have contributed to the population dynamics and distribution of a widespread terrestrial mammal, and how these processes may have helped shape the level and pattern of genetic diversity in modern populations.
Our study species is a large mammal that is currently the most common and widespread cervid in temperate habitat across Europe, the European roe deer (Capreolus capreolus; see Andersen et al., 1998). The European roe deer is well represented in the fossil record from the Middle to Late Pleistocene (Lister et al., 1998; Sommer and Zachos, 2009), and have coexisted with many other large mammal species that perished around the Pleistocene/Holocene transition (for example, mammoth, cave bear, steppe bison, giant deer). During the last glacial maximum (LGM, 23 000–18 000 years before present (YBP); Kukla et al., 2002), roe deer were likely forced into southern refugial populations along with other temperate species, later recolonising northern Europe following climatic warming and deglaciation.
Signatures of this past history are evident in the phylogeographic patterns of modern roe deer from across Europe (for review, see Sommer et al., 2009). Genetic variation of the European roe deer divides into central, eastern and western lineages. The central lineage is widespread throughout Europe, whereas the eastern lineage is found mainly in Greece and Serbia and the western lineage is mainly in Spain and Portugal (see Lorenzini et al., 2003; Randi et al., 2004; Lorenzini and Lovari, 2006). In addition to these divisions, significant internal structuring has been detected in roe sampled from the Italian and Iberian peninsulas. This supports the existence of a subspecies in central–southern Italy (C. c. italicus) (Lorenzini et al., 2002; Randi et al., 2004) and an additional Celtic–Iberian group in central–southern Spain (Royo et al., 2007). Taken together, these data likely reflect the existence of several glacial refugia (Sommer et al., 2009). Following the end of the LGM, roe deer recolonised Britain via the expanse of land known as Doggerland that once provided a direct connection to continental Europe (Yalden, 1999).
Although climatic change has been a major force shaping the evolutionary history of the roe deer, in modern times the ever-growing impact of humans has also been significant (Baker and Hoelzel, 2013). Cervids across Europe have long been under the influence of humans through hunting, habitat modifications and restocking. In Britain, it was not until the last few centuries (fourteenth–eighteenth) that the impact of human activity through hunting and deforestation began to significantly affect populations of the British roe deer. These activities were believed to have caused local extinction in most areas of southern United Kingdom (Ritson, 1933), and restricted roe populations to parts of Scotland and possibly some of the northern border English counties (Whitehead 1964). With the turn of the nineteenth century, large-scale replanting of woodland provided suitable habitat for remnant populations over much of the north to recolonise uninhabited areas (Taylor, 1948). In the south, reforestation helped facilitate the successful reintroduction of roe deer (using both native and nonnative stocks) across much of southern United Kingdom (Whitehead, 1964; Prior, 1995; Baker and Hoelzel, 2013). Reforestation and reintroductions have been so successful that roe deer populations are showing continual expansion and repopulation over much of their historic range (Whitehead, 1964; Ward, 2005). A recent study suggested that genetic structure and diversity of British roe deer populations has been strongly influenced by recent bottlenecks and restocking activity, as well as specific aspects of life history and behaviour (Baker and Hoelzel, 2013).
In this study we focus on the British population using both ancient and modern DNA, and integrate these data into published studies on roe deer genetic diversity across Europe. We test the hypothesis that historical founder and colonisation events (both natural and anthropogenic) were instrumental in defining modern patterns of diversity and population structure in this species. We also test the hypothesis that there was free movement across the land bridge at Doggerland until the land bridge was flooded, isolating two established populations either side of the British Channel. Our results provide inference about the role of historical demographic events on the modern genetic diversity of a widespread species, and reinforce earlier inference about the role of life history characteristics (Baker and Hoelzel, 2013).
Materials and methods
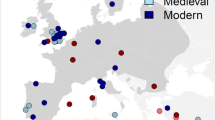
Ancient samples (N=140) were collected from across the United Kingdom (Table 1 and Supplementary Table S1). Samples that showed amplification success are represented on a map of Britain (N=86; Figure 1 and Supplementary Table S1). DNA was extracted from 0.05 g of bone powder using a QIAquick purification kit (Qiagen, Hilden, Germany) following the manufacturer’s guidelines. Precautions to avoid contamination were taken during every stage of ancient DNA (aDNA) extraction and PCR set-up that took place in a separate laboratory dedicated to aDNA research free from contemporary DNA or PCR product. No laboratory materials or clothing were transferred from the postamplification rooms to the ancient laboratory. All work surfaces and equipment were thoroughly cleaned with 10% bleach (sodium hypochlorite) followed by 70% ethanol. Surfaces, equipment and solutions were also routinely exposed to ultraviolet light for at least 10 min. All extractions and PCR work were carried out in class II PCR hoods. Negative extraction and PCR controls (1 sample in every 5) were included to detect potential contamination in reagents and cross-contamination between samples. In addition, 50% of samples were replicated by extracting twice from independent samples of the same bone. In addition, all samples with unique aDNA haplotypes were repeated with an independent PCR amplification. DNA sequences (from both independent DNA extractions and PCRs) were considered authentic when independent replicates from the same individual yielded identical sequences. In rare cases when consistent differences between any replicates were detected, a third replicate was used to verify the actual nucleotide position.

Map showing the locations from where modern (black circles) and ancient (grey circles) mtDNA sequences originated from. The sample numbers are shown either alongside or within each sample circle.
Two overlapping fragments of the hypervariable section of the mitochondrial control region were designed to overlap with database modern sequences. The primer pairs were Roe_1F: 5′-ATTATATGCCCCATGCTTAT-3′ and Roe_1R: 5′-CCTGAAGAAAGAACCAGATG-3′; Roe_2F: 5′-AACCAAGAACTTTACCAG-3′ and Roe_2R: 5′-GGGACATAATGTACTATG-3′. These primer pairs amplified fragments of 244 and 267 bp, respectively (including primers). PCR Reactions (25 μl) contained 0.2 pM μl−1 each primer, 0.2 mM of each dNTP, Platinum 1X High Fidelity Buffer (60 mM Tris-SO4, pH 8.9/18 mM (NH4)2SO4 (Invitrogen)), 1.5 mM MgCl2 and 1 unit of Taq High Fidelity DNA polymerase (Invitrogen). Then, 2 μl of DNA template was added. Amplifications were performed with the following cycles: 95 °C for 5 min; 45 cycles at 94 °C for 45 s, 51 °C (roe_1F, 1R) or 55 °C (roe_2F, 2R) for 45 s and 68 °C for 45 s; 68 °C for 5 min. Amplicons were sequenced in both directions and PCR products were cleaned using the QIAquick PCR Purification Kit (Qiagen) according to the manufacturer’s instructions.
Phylogeography, diversity and expansion signals
Our aDNA sequence data were compared against three published modern data sets (Randi et al., 2004; Royo et al., 2007; Baker and Hoelzel, 2013). Details of the exact data used and corresponding accession numbers are provided in Supplementary Table S4. All sequences were aligned against each other using the programme CLUSTAL X (Larkin et al., 2007). A 388-bp region of the consensus mitochondrial DNA (mtDNA) control region was used for subsequent analyses. The European data set was divided into the lineages eastern, western and central as well as the subspecies C. c. italicus (as previously described by Randi et al., 2004) and the Celtic–Iberian group (as previously described by Royo et al., 2007).
Summary statistics were calculated in DnaSP10.4.9 (Rozas et al., 2003) for each of the lineages individually and combined, as well as for the ancient and modern UK samples individually and combined. The following statistics were computed: number of segregating (polymorphic) sites (S); number of unique haplotypes (h); haplotype diversity (H); average number of pairwise nucleotide differences (k); and nucleotide diversity (π).
The relationship between European and UK populations (using both ancient and modern data) was investigated using a Median Joining Network constructed using the programme NETWORK 4.5 (http://www.fluxus-engineering.com), chosen to account for possible missing nodes and alternative connections (Bandelt et al., 1999). The FST and ΦST values were calculated using Arlequin v 3.0 (Excoffier et al., 2005). Two neutrality tests were performed in DnaSP: Tajima’s D (Tajima, 1989) and Fu’s Fs (Fu, 1997). Mismatch distributions were also used to evaluate possible events of expansion and decline (Rogers and Harpending, 1992) using the sudden expansion model and goodness-of-fit tests (sum of squared deviation (SSD); Harpending’s raggedness index R; Schneider and Excoffier, 1999). Tau (τ; calculated using ARLEQUIN 3.5) was used to estimate expansion time (T) using the equation: T=τ/2 μ where μ is the mutation rate in units of substitutions per locus per generation (Rogers and Harpending, 1992). The roe deer generation time was taken to be 3 years (after Randi et al., 1998, 2004).
Demographic analyses
Given that our UK ancient sample set reflected a substantial sample size, covered a time frame of over 5000 years and was less likely to be affected by continental introductions, all demographic analyses were based on just these samples. The programme BEAST v 1.4.8 (Drummond and Rambaut, 2007) was used to obtain substitution parameters (based on the ancient sample set) and explore past demographic change in roe deer. Substitution rates were estimated from temporally spaced sequence data (Drummond et al., 2002) obtained by stratigraphic dating. For some samples, stratigraphic dates had wide date ranges and therefore an average date was used across all samples. All dates were provided as YBP. Input files were first generated with BEAUTi version 1.4.2. (Drummond and Rambaut, 2007).
Three independent Markov chain Monte Carlo runs of four chains each were run for 20 000 000 iterations, of which the first 10% were discarded as burn-in. Samples from two runs (that yielded similar results) were combined to estimate model parameters. Genealogies and model parameters were sampled every 2000 iterations. An explicit post-mortem damage model was incorporated into each run (Rambaut et al., 2009) that takes into account the potential for sequence damage to influence the outcome of the aDNA analyses. Demographic inferences were essentially the same with or without the incorporation of the post-mortem damage model (details available from authors). A strict molecular clock model was applied. To determine the model of sequence evolution to use in this programme, a hierarchical likelihood test in Mr. MODELTEST 2.2 (Posada and Crandall, 1998) was performed. The substitution model chosen was Hasegawa, Kishino and Yano (HKY) (Hasegawa et al., 1985). Independent runs were combined using Tracer 1.4 (Rambaut and Drummond, 2007) to generate credibility intervals that represent the coalescent model and phylogenetic uncertainty and to produce final estimates. For combined runs, effective sample sizes for each parameter exceeded 100, which indicated efficient mixing (that is, low autocorrelation in the Markov chain) and sufficient sampling of model parameters. The Bayesian skyline plot (BSP) demographic model was applied (Drummond et al., 2005).
The programme isolation with migration (IM; Hey and Nielsen, 2004) was used to estimate divergence times between the ancient UK population and its most closely related European populations. To minimise substructure within sample sets, the mainland European data set was restricted to sequences from central Europe (Germany and France), resulting in a comparison between 37 central European and 86 ancient UK sequences. The substitution rate estimated from BEAST was incorporated and the HKY model of evolution applied. Three runs were conducted using a two-step heating increment. Each Markov chain was run for 100 000 000 generations after discarding 10% burn-in. The first run was conducted to determine appropriate priors for subsequent runs; unrealistic upper bounds for priors were used in this preliminary run. Uninformative priors (that is, ranges that encompassed the entire posterior distributions) were then set for the final two runs. The final runs were conducted using identical conditions but different random number seeds to test whether multiple runs gave similar results. To ensure convergence, simulations were run until the smallest effective sample size estimates were at least 100 (Hey, 2005). Results from replicate runs did not differ, and hence data from the longer of the two runs are presented. The mode is reported along with the 95% highest posterior densities (HPDs; Hey and Nielsen, 2004).
Direct comparison between ancient and modern populations
To compare phylogenies between modern and ancient haplotypes, median joining networks were created in NETWORK 4.5 (Bandelt et al., 1999). Inferences about possible native and nonnative haplotypes were made by mapping haplotypes common to ancient and modern populations. To investigate relationships between ancient and modern populations, pairwise FST values (Weir and Cockerham, 1984) were calculated for mtDNA with 1000 permutations in Arlequin 3.0 (Excoffier et al., 2005). For the calculation of FST values, the ancient population could only be divided as north and south because of restrictions of small sample sizes per area, whereas the modern populations were separated by area into six different populations. Bonferroni correction was applied to correct for type 1 error in multiple tests.
Results
Phylogeography, genetic diversity and expansion signals
The data taken from Randi et al. (2004) and Royo et al. (2007) were divided into the following five separate groups (thought to reflect historical geographic refugial populations): an eastern, western, central, and Celtic–Iberian groups and the C. c. italicus subspecies. With the addition of UK samples, this structure was maintained (Figure 2). The inclusion of UK data increased the number of haplotypes found across Europe from 95 (see Royo et al., 2007) to 115. Some of the haplotypes found in both ancient and modern UK populations were shared with the central European lineage, indicating a close association (Figure 2). The FST and ΦST values were used to quantify the degree of differentiation among these lineages and in comparison with UK data (Table 1; Randi et al., 2004).

Median joining network (MJN) computed using 115 haplotypes from continental European data sets along with the ancient and modern UK data sets. Circle size is proportional to haplotype frequencies. The shaded circles represent the lineages, subspecies and groups formerly defined by Randi et al. (2004) and Royo et al. (2007). A full colour version of this figure is available at the Heredity journal online where the central lineage is yellow, eastern lineage is green, western lineage is blue, the subspecies C. c. italicus is white, and the Celtic–Iberian group is purple. For the UK populations from this study, ancient UK is grey and modern UK is black.
Overall, levels of mtDNA diversity showed a wide range within Europe (see Table 2). The central European lineage had the highest levels of diversity and the subspecies C. c. italicus the lowest. Diversity in ancient UK populations was similar to the main European lineages, whereas modern UK populations were less diverse. Fu’s Fs indicated a significant departure from neutrality, indicating expansion for the central lineage all European lineages combined with C. c. Italicus and ancient UK and modern and ancient UK data combined (Table 2). The more conservative Tajima’s D test indicated negative but nonsignificant deviations from neutrality for these same sample sets (Table 2).
The goodness-of-fit tests comparing an expansion model to the observed mismatch distributions revealed strong evidence for expansion for all European lineages combined with C.c. Italicus (SSD=0.003, P=0.41; R=0.012, P=0.47), the ancient UK population (SSD=0.007, P=0.68; R=0.036, P=0.34), modern and ancient UK combined (SSD=0.010, P=0.28; R=0.035. P=0.46) but not for the central lineage (SSD=0.019, P=0.00; R=0.089, P=0.00).
Mismatch analyses, from which τ was calculated, are represented in Figures 3a–d. Possible expansion events occurred at τ=3.08 (7014 YBP; 95% HPD 7919–3932YBP) for the central lineage and at τ=5.31 (12 069 YBP; 95% HPD 19 479–4984) for all European lineages combined with C. c. Italicus. For ancient UK populations the data indicate a possible expansion event at τ=2.76 (6274 YBP; 95% HPD 10 273–2072 YBP).

Mismatch distributions for mitochondrial DNA haplotypes sampled from the central lineage (a), all European lineages and C. c. italicus combined (b), ancient UK (c) and ancient and modern UK populations combined (d).
Demographic analyses
The substitution rate estimated by BSP in BEAST was 3.69 × 10−7 (95% HPD interval 1.82 × 10−7 to 5.82 × 10−7) substitutions per site per year (Supplementary Figure S1). A coalescent reconstruction of past population dynamics (BSP) of British roe deer based on aDNA shows a rapid expansion in the effective number of roe deer between 5000 and 6000 YBP. After that time frame, roe deer numbers appear to remain relatively stable (Figure 4).

A Bayesian skyline plot derived from ancient UK roe deer mtDNA D-loop sequences. The x axis is in units of years before present, and the y axis is equal to population size (the product of the effective population size and the generation length in years: 3). The black line is the median estimate, and the blue area shows the 95% highest posterior density intervals.
The divergence time for ancient UK and central Europe calculated in IMa is well resolved (Table 3 and Supplementary Figure S2), with a posterior distribution that has a distinct peak and bounds that fall within the prior distribution. Comparing ancient UK and the central European populations, the position of the peak indicates a population split at 5369 YBP (95% HPD 3317–12 901 YBP; Table 3). The effective population size for ancient United Kingdom is estimated to be largest, but central Europe and ancient United Kingdom are similar (with overlapping confidence interval ranges). The estimated rate of mtDNA gene flow into the United Kingdom and Europe is very small, suggesting that after separation the populations remained isolated.
Direct comparison between ancient and modern DNA
Between ancient and modern periods, haplotype number, haplotype diversity, nucleotide diversity and k decreased (Figure 5 and Table 2). We successfully sequenced 86 (61.4%) of the ancient UK samples (see Table 1 and Supplementary Table S1, for details) from which we identified 24 haplotypes (h/N=0.279). From our 279 modern UK samples, 12 haplotypes were detected (Figure 5 and Table 2; h/N=0.043). Six haplotypes were common to both ancient and modern populations and most of these occurred in northern United Kingdom. Some haplotypes common to both time points were sampled at very low frequencies and found in isolated populations (for example, m7, m8 and m11). Six haplotypes, unique only to modern populations, may represent haplotypes that have either gone undetected in historical populations or that have been introduced from nonnative locations. One population, which likely reflects the latter scenario, is that of Norfolk, which is known to be a site of nonnative reintroduction, and distinctly exhibits the single unique haplotype m3 (Figure 5b and Table 3). Genetic differentiation between all ancient and UK contemporary populations was significant based on mitochondrial data (Table 4). The most closely related contemporary population to ancient UK populations was Lancashire based on mitochondrial data (the only nonsignificant difference). In general, mitochondrial data showed that contemporary populations found in the north were more closely related to ancient UK populations than those found in the south. Specifically, the southern population of Norfolk was most distantly related to ancient populations.

A direct comparison of ancient (a) versus modern (b) median joining networks computed in NETWORK. In (a), grey circles indicate ancient haplotypes (n=24; a1–24; see Supplementary Table S2). In (b), black circles indicate modern haplotypes (n=12; m1–12; see Supplementary Table S3). For both networks, the asterisks (*) denote those haplotypes that are common to both ancient and modern populations.
Discussion
Phylogeography, genetic diversity and expansion signals
Based on a 750-bp region of the mtDNA, Randi et al. (2004) identified the existence of three main roe deer lineages in Europe; central, eastern and western together with the subspecies C. c. italicus, which each likely represent independent glacial refugia. Royo et al. (2007) later described the existence of a Celtic–Iberian group. When the UK data were combined with these European data sets, and a reduced consensus region (388 bp) examined, the same structure was maintained (Figure 2). The inclusion of the UK ancient and modern genetic data revealed that 22 haplotypes were unique to Britain. The finding that all UK haplotypes cluster with the central lineage strongly supports its origins from this lineage (see Table 1 and Figure 2). Colonisation from northern Europe near the land bridge, where the central lineage is common, would be expected based on geographic proximity.
The location of the refuge from which the central lineage originated is still unknown, although a Carpathian or further eastern origin is supported by recent molecular and fossil data (see Randi et al., 2004; Lorenzini and Lovari, 2006; Sommer and Zachos, 2009). A colonisation route from this area would correlate well with patterns of broad-leaf forest expansion (Petit et al., 2003) that is the preferred habitat of the roe deer (Putman and Langbein, 2003). Our comparisons shown in Table 1 support the affinity between the UK samples and the central lineage illustrated in the network (Figure 2), and this seems strongest for the ancient sample set. A stronger association between the ancient UK and modern European sample sets may reflect the greater inclusion of samples from translocated and bottlenecked populations among the modern UK samples. Although studies based exclusively on mtDNA data run the risk of misinterpretation because of looking only at matriline history or only a single gene tree, roe deer comparative analyses tend to show good agreement between nuclear and mtDNA indications of population structure, as seen both on a fine geographic scale in the United Kingdom (Baker and Hoelzel, 2013) and on a broader European scale (Randi et al., 2004; Lorenzini and Lovari, 2006; Sommer and Zachos, 2009). As of yet, there are no nuclear DNA-based studies that compare roe deer populations in the United Kingdom with European populations.
Both neutrality tests and mismatch distributions suggested strong expansion events for all European lineages combined, the ancient UK sample alone, and the ancient and modern UK samples combined. Calculating tau (τ) from the mismatch distributions and using the substitution rate calculated in BEAST, there was evidence of expansion at 13 500 YBP for the combined European lineage, and 6300 YBP for the UK population. The expansion date for the UK population was consistent with the BSP graph that showed that UK roe populations expanded over a similar timescale (Figure 4).
Mismatch analyses detected possible expansion events (or selective sweeps) for roe deer following the LGM (see Figure 3). The LGM (23 000–18 000 YBP; Kukla et al., 2002) confined roe and other temperate species to separate southern glacial refugia. This was because of the permafrost and Arctic tundra ecosystems that were widespread in central Europe down to a latitude of 45° (Andersen and Borns, 1997). Following the LGM, species were able to recolonise by expansion into formerly glaciated regions. According to the fossil record, it was not until the period of warming, between 14 700 and 11 600 YBP, that roe deer were able to rapidly expand into much of Europe (Sommer et al., 2009; Sommer and Zachos, 2009). This is consistent with the estimated expansion date for the European lineages (12 000 YBP; Figure 3).
For northern European lowlands (such as the United Kingdom), fossil evidence suggests that roe did not recolonise until the early Holocene (Sommer and Zachos, 2009). For the United Kingdom, the earliest evidence for postglacial recolonisation originates from Thatcham in southern England where bones were radiocarbon dated to 9439±100 YBP (Sommer et al., 2009). During this time, the central European lowlands were apparently being slowly recolonised by birch and pine woods (Usinger, 2004) that would have in turn improved the environmental conditions for roe deer. However, it was not until 6000 YBP that the vegetation pattern broadly resembled that of today (Hewitt, 1999), and the expansion signal for ancient UK roe populations at 6300 YBP (Figure 3) may have been a response to the improved environmental conditions.
Taken together, our results indicate that new habitat was quickly exploited by expanding roe deer populations following the end of the last glaciation. Randi et al. (2004) previously proposed two expansion events for continental European populations (based on 750-bp segments of the mtDNA control region), scaling values of τ from their mismatch analyses with a ‘phylogenetic rate’ of 4–6% per million years. The resulting expansion times were estimated to have coincided with the penultimate (c. 250 Ka) and the last (c. 130 Ka) interglacials. Fossil evidence suggests that roe deer have been present in Europe through at least 600 000 years, since the Middle Pleistocene (Lister et al., 1998). Using the same data but with the new substitution rate calculated in this study (37% per million years), it is estimated that expansions instead occurred at 13 300 (HPD; 8400–22 900) YBP and 8400 YBP (HPD; 5300–17 100), respectively. Both of these expansion dates are consistent with the European expansion signals proposed above, based on the same data, but with a reduced sequence length (388 bp).
The results of this study are consistent with a number of recent studies that have replaced ‘phylogenetic rates’ with substitution rates directly calibrated using aDNA (Ho et al., 2005), more relevant to the shorter time frame of the Holocene. For example, the substitution rate from our study (3.69 × 10−7 s.s.yr −1) is similar to that for the ancient brown bear (Saarma et al., 2007) and bison (Shapiro et al., 2004) (3.2 × 10−7 and 3.0 × 10−7 s.s.yr−1, respectively). In fact, reviews show broad consistency among a wide range of studies (Ho et al., 2005, 2008). It is unlikely that all aDNA data sets are confounded by error, as these errors would need to have been made systematically and substantially (Ho et al., 2007), The use of the roe deer substitution rate that we calculated suggested that divergence and population expansions occur over much shorter timescales than previously proposed. As for previous studies, the more recent dates (based on higher substitution rates) are consistent with expectations based on historical environmental events (see de Bruyn et al., 2011). For the roe deer in Europe, the expansion date is consistent with expected postglacial expansions. For the UK sample, it is consistent with the separation of the United Kingdom from the European landmass.
Demographic analyses
For the UK sample, expansion signals from IM, mismatch distributions and the BSP plot are around the time of the separation of the United Kingdom from the European landmass. When the Scandinavian and British ice sheets reached their maximum extent, and the North Sea as a consequence receded to its lowest level, Britain was connected to the continent by a land bridge (Fairbanks, 1989). This dry land, referred to as Doggerland, would have allowed roe deer to migrate from central Europe. Doggerland likely existed between 8000 (Shennan et al., 2000; Sturt et al., 2013) and 5800 YBP (van der Molen and van Dijck, 2000) when its loss resulted from warming, unlocking large quantities of water from ice caps and causing sea levels to rise. A channel restricting movement likely developed early in this period. The split may have coincided with increasingly suitable habitat in the United Kingdom, resulting in expansion of a population recently isolated from the source population in Europe (Sommer and Zachos, 2009).
Direct comparison between ancient and modern DNA
Direct comparisons between ancient and modern populations in the United Kingdom revealed an overall loss in genetic variation (see Table 2 and Figure 5) that could be attributed to a recent period of bottlenecking caused by overhunting and deforestation between the late fourteenth and eighteenth centuries (Whitehead, 1964), followed by the establishment of new populations based on small founder groups. Conservation biology is often concerned with preserving native populations to secure large-scale genetic diversity and preserve possible local adaptations (Nielsen et al., 1999). In this respect, it appears that populations in the most northern parts of the United Kingdom (for example, Scotland; Perth/Moray/Glasgow) would be most important to conserve. These populations have retained high number of ‘native’ haplotypes (that is, those present in historical populations; see Figure 5 and Supplementary Table S3), show a close relationship with historical populations (Table 4) and exhibit high levels of both microsatellite DNA and mtDNA variability relative to other populations (see Baker and Hoelzel, 2013).
This result is concordant with the historical record that suggested that medieval bottlenecking was less intense in Scotland. For example, Whitehead (1964) claimed that roe never went extinct in this region and may have retained appreciable numbers. The other northern populations, such as those in northern England (for example, Carlisle, Durham, North York, Lancashire), appear to be primarily native, but lower representative numbers of haplotypes (see Supplementary Table S3) may reflect near extinction or extinction in these areas followed by later re-establishment through expansion (see Baker and Hoelzel, 2013). This inference is also supported by the historical record (Bewick, 1790). Other areas of the United Kingdom that appear to harbour populations based on native stock include extant southern populations (for example, Dorset, Wiltshire, Somerset, Berks) that were founded from stock translocated from Scotland (Whitehead, 1964) after the medieval bottleneck that extirpated the original southern populations. The genetic record is concordant with this scenario, as it appears that large losses of haplotypes between ancient and modern periods have occurred in the south (see Supplementary Tables S2 and S3) and that the three native haplotypes currently present in these southern populations (m1, m2 and m4; see Figure 4) are also common to Scotland (see Supplementary Table S3).
For other areas of the United Kingdom, it appears that populations have experienced some level of influence of restocking involving nonnative individuals. The Lancashire population was characterised by three native haplotypes (m1, m4, m6; see Supplementary Table S3) and one potentially nonnative introduced haplotype (that is, absent from ancient populations; see Figure 4). The latter nonnative haplotype may reflect the relicts of an introduction event that occurred when 12 Austrian roe were introduced into this population in 1913 to ‘improve the local breed’ (Whitehead, 1964; Prior, 1995), although it is also possible that it was present in the ancient population, but not detected. There was evidence for complete lineage replacement in Norfolk, a finding that is consistent with the records of the human translocation of nonnative (German) stock into this area (see Whitehead, 1964). This was supported by the findings that a single, novel haplotype unique to this location (m3; Figure 3) was detected and that this population exhibited the highest levels of differentiation (based on both mitochondrial (see Table 4) and microsatellite FST values; see Baker and Hoelzel, 2013) when compared with ancient and other contemporary UK populations.
Conclusion
This study has used ancient and modern DNA to provide otherwise intractable information on the evolutionary events that have shaped European roe deer during its recent postglacial history, including direct data on the impact of a major vicariance event (the closing of the land bridge). Recolonisation of the roe deer across Europe seems to have occurred very rapidly as environmental conditions improved following the end of the LGM, and our data on this process agree well with inference from the fossil records. Shortly after, roe deer recolonised the United Kingdom but populations became isolated as the land bridge was cut from mainland Europe. Our data provide a plausible time frame for when this occurred, and suggest a postisolation expansion. More recent anthropogenic events led to a pattern of historical bottlenecks and reintroductions, and for these events the genetic data track well with the historical data. Since establishment during the Holocene, British roe deer populations have evolved considerable genetic structure (see Baker and Hoelzel, 2013), reflecting processes associated with both natural environmental and anthropogenic changes. Understanding the integration of specific responses to climatic and anthropogenic change in species of conservation concern can help predict future patterns of diversity (Hadly and Barnosky, 2009) that may be fundamental to long-term conservation and species management planning (Leonard, 2008).
Data archiving
Sequence data have been submitted to GenBank: accession numbers JX971589–JX971615; KF961012–KF961035.
Accession codes
References
Andersen BG, Borns HW . (1997) The Ice Age world: An Introduction to Quatenary History and Research with Emphasis on North America and Europe During the Last 2.5 Million Years. Scandinavian University Press: Oslo.
Andersen R, Duncan P, Linnell JDC . (1998) The European Roe Deer: The Biology of Success. Scandinavian University Press: Oslo.
Baker KH, Hoelzel AR . (2013). Evolution of population genetic structure of the British roe deer by natural and anthropogenic processes (Capreolus capreolus). Ecol evol 3: 89–102.
Bandelt HJ, Forster P, Rohl A . (1999). Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16: 37–48.
Bewick T . (1790) A General History of the Quadrupeds. Hodgson, Beilby and Bewick: Newcastle upon-Tyne.
de Bruyn M, Hoelzel AR, Carvalho GR, Hofreiter M . (2011). Faunal histories from Holocene ancient DNA. Trends Ecol Evol 26: 405–413.
Drummond AJ, Nicholls GK, Rodrigo AG, Solomon W . (2002). Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161: 1307–1320.
Drummond AJ, Rambaut A . (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7: 214.
Drummond AJ, Rambaut A, Shapiro B, Pybus OG . (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22: 1185–1192.
Excoffier L, Laval G, Schneider S . (2005). Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1: 47–50.
Fairbanks RG . (1989). A 17000-year glacio-eustatic sea level record: influence of glacial melting rates on the Younger Dryas event and deep ocean circulation. Nature 342: 637–642.
Fu YX . (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925.
Hadly EA, Barnosky AD . (2009). Vertebrate fossils and the future of conservation biology. Paeon Soc Papers 15: 39–59.
Hasegawa M, Kishino H, Yano TA . (1985). Dating the human ape spliiting by a molecular clock of mitochondrial-DNA. J Mol Evol 22: 160–174.
Hewitt GM . (1999). Post-glacial re-colonization of European biota. Biol J Linn Soc 68: 87–112.
Hey J . (2005). On the number of New World founders: a population genetic portrait of the peopling of the Americas. PLoS Biol 3: 965–975.
Hey J, Nielsen R . (2004). Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167: 747–760.
Ho SYW, Kolokotronis SO, Allaby RG . (2007). Elevated substitution rates estimated from ancient DNA sequences. Biol Lett 3: 702–705.
Ho SYW, Phillips MJ, Cooper A, Drummond AJ . (2005). Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol Biol Evol 22: 1561–1568.
Ho SYW, Saarma U, Barnett R, Haile J, Shapiro B . (2008). The effect of inappropriate calibration: three case studies in molecular ecology. PLoS One 3: e1615.
Kukla GJ, Clement AC, Cane MA, Gavin JE, Zebiak SE . (2002). Last interglacial and early glacial ENSO. Quatern Res 58: 27–31.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23: 2947–2948.
Leonard JA . (2008). Ancient DNA applications for wildlife conservation. Mol Ecol 17: 4186–4196.
Lister AM, Grubb P, Sumner SRM . (1998). Taxonomy, morphology and evolution of European roe deer. In: Duncan P, Linell JDC, Andersen R (eds). The European Roe Deer: The Biology of Success. Scandinavian University Press: Oslo, Norway. pp 23–46.
Lorenzini R, Lovari S . (2006). Genetic diversity and phylogeography of the European roe deer: the refuge area theory revisited. Biol J Linn Soc 88: 85–100.
Lorenzini R, Lovari S, Masseti M . (2002). The rediscovery of the Italian roe deer: genetic differentiation and management implications. Ital J Zool 69: 367–379.
Lorenzini R, San Jose C, Braza F, Aragon S . (2003). Genetic differentiation and phylogeography of roe deer in Spain, as suggested by mitochondrial DNA and microsatellite analysis. Ital J Zool 70: 89–99.
Nielsen EE, Hansen MM, Loeschcke V . (1999). Analysis of DNA from old scale samples: technical aspects, applications and perspectives for conservation. Hereditas 130: 265–276.
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R et al. (2003). Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300: 1563–1565.
Posada D, Crandall KA . (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14: 817–818.
Prior R . (1995) The Roe Deer: Conservation of a Native Species. Swan Hill Press.
Putman R, Langbein J . (2003) The Deer Manager's Companion; A Guide to the Management of Deer in the Wild and in Parks. Swan Hill Press: Shrewsbury.
Rambaut A, Drummond AJ . (2007) Tracer v 1.4. University of Edinburgh: UK.
Rambaut A, Ho SYW, Drummond AJ, Shapiro B . (2009). Accommodating the effect of ancient DNA damage on inferences of demographic histories. Mol Biol Evol 26: 245–248.
Randi E, Alves PC, Carranza J, Milosevic-Zlatanovic S, Sfougaris A, Mucci N . (2004). Phylogeography of roe deer (Capreolus capreolus) populations: the effects of historical genetic subdivisions and recent nonequilibrium dynamics. Mol Ecol 13: 3071–3083.
Randi E, Pierpaoli M, Danilkin A . (1998). Mitochondrial DNA polymorphism in populations of Siberian and European roe deer (Capreolus pygargus and Capreolus capreolus). Heredity 80: 429–437.
Ritson G . (1933) The Roe Deer in Cumberland. Natural History Society: Carlisle.
Rogers AR, Harpending H . (1992). Population growth makes waves in the distribution of pairwise genetic- differences. Mol Biol Evol 9: 552–569.
Royo LJ, Pajares G, Alvarez I, Fernandez I, Goyache F . (2007). Genetic variability and differentiation in Spanish roe deer (Capreolus capreolus): a phylogeographic reassessment within the European framework. Mol Phylogenet Evol 42: 47–61.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R . (2003). DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497.
Saarma U, Ho SYW, Pybus OG, Kaljuste M, Tumanov IL, Kojola I et al. (2007). Mitogenetic structure of brown bears (Ursus arctos) in northeastern Europe and a new time frame for the formation of European brown bear lineages. Mol Ecol 16: 401–413.
Schneider S, Excoffier L . (1999). Estimation of past demographic parameters from the distribution of pairwise differences when the mutation rates very among sites: application to human mitochondrial DNA. Genetics 152: 1079–1089.
Shapiro B, Drummond AJ, Rambaut A, Wilson MC, Matheus PE, Sher AV et al. (2004). Rise and fall of the Beringian steppe bison. Science 306: 1561–1565.
Shennan I, Lambeck K, Flather R, Horton B, McCartur J, Innes J et al. (2000). Modelling western North Sea palaeogeographies and tidal changes during the Holocene. In: Shennan I, Andrews J (eds). Holocene Land-Ocean Interaction and Environmental Change around the North Sea Vol 166, Geological Society Special publications: London. pp 299–319.
Sommer RS, Fahlke JM, Schmolcke U, Benecke N, Zachos FE . (2009). Quaternary history of the European roe deer Capreolus capreolus. Mammal Rev 39: 1–16.
Sommer RS, Zachos FE . (2009). Fossil evidence and phylogeography of temperate species: ‘glacial refugia’ and post-glacial recolonization. J Biogeogr 36: 2013–2020.
Sturt F, Garrow D, Bradley S . (2013). New models of North West European Holocene palaeogeography and inundation. J Archaeol Sci 40: 3963–3976.
Tajima F . (1989). The effect of change in population size on DNA polymorphism. Genetics 123: 597–601.
Taylor WL . (1948). The distribution of wild deer in England and Wales. J Anim Ecol 17: 151–164.
Usinger H . (2004). Vegetation and climate of the lowlands of northern Central Europe and adjacent areas around the Younger Dryas–Preboreal transition–with special emphasis on the Preboreal oscillation. Hunters in a Changing World. In: Terberger T, Eriksen BV (eds). Environment and Archaeology of the Pleistocene-Holocene Transition (ca. 11 000–9000 B.C.) in Northern Europe. Marie Leidorf Publisher: Rahden. pp 1–26.
van der Molen J, van Dijck B . (2000). The evolution of the Dutch and Belgian coasts and the role of sand supply from the North Sea. Global Planet Change 27: 223–244.
Ward AI . (2005). Expanding ranges of wild and feral deer in Great Britain. Mammal Rev 35: 165–173.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Whitehead GK . (1964) The Deer of Great Britain and Ireland. Routledge & Kegan Paul: London.
Yalden DW . (1999) The History of British Mammals. Poyser: London.
Acknowledgements
We thank Dr Umberto Albarella at the University of Sheffield, Professor Andrew Chamberlain at the University of Manchester, Dr Naomi Sykes at the University of Nottingham, Derek Hurst at Worcestershire Archaeological Service, Erica Macey at Birmingham Archaeology, Richard Sabin at the Natural History Museum, Peta Sadler, Huw Sherlock at Archenfield Archaeology, Catherine Smith at Scottish Urban Archaeological Trust, Ian Smith at Chester Archaeology Services, Roy Stephenson at MOLAS and, finally, Dr Rob Symmons at Fishbourne Roman Palace for the provision of ancient roe deer samples. We thank all deer stalkers and managers who helped collect modern roe deer samples as well as Hugh Rose who helped coordinate this process. Finally, we thank the British Deer Society and the Kenneth Whitehead Trust for funding KHB in this PhD studentship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Baker, K., Hoelzel, A. Influence of Holocene environmental change and anthropogenic impact on the diversity and distribution of roe deer. Heredity 112, 607–615 (2014). https://doi.org/10.1038/hdy.2013.142
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2013.142
Keywords
This article is cited by
-
Mitochondrial DNA diversity and the population genetic structure of contemporary roe deer (Capreolus capreolus) in Europe
Mammalian Biology (2022)
-
Eco-Evolutionary Processes Generating Diversity Among Bottlenose Dolphin, Tursiops truncatus, Populations off Baja California, Mexico
Evolutionary Biology (2018)
