
Abstract
Mating processes of local demes and spatial genetic structure of island populations at the self-incompatibility (S-) locus under negative frequency-dependent selection (NFDS) were evaluated in Prunus lannesiana var. speciosa in comparison with nuclear simple sequence repeat (SSR) loci that seemed to be evolutionarily neutral. Our observations of local mating patterns indicated that male–female pair fecundity was influenced by not only self-incompatibility, but also various factors, such as kinship, pollen production and flowering synchrony. In spite of the mating bias caused by these factors, the NFDS effect on changes in allele frequencies from potential mates to mating pollen was detected at the S-locus but not at the SSR loci, although the changes from adult to juvenile cohorts were not apparent at any loci. Genetic differentiation and isolation-by-distance over various spatial scales were smaller at the S-locus than at the SSR loci, as expected under the NFDS. Allele-sharing distributions among the populations also had a unimodal pattern at the S-locus, indicating the NFDS effect except for alleles unique to individual populations probably due to isolation among islands, although this pattern was not exhibited by the SSR loci. Our results suggest that the NFDS at the S-locus has an impact on both the mating patterns and the genetic structure in the P. lannesiana populations studied.
Similar content being viewed by others

Introduction
Homomorphic self-incompatibility (SI) systems are widespread physiological mechanisms that prevent self-fertilization in angiosperms by controlling pollen germination or pollen tube growth (de Nettancourt, 1977). Recognition between the pollen and the pistil involves specificity molecules, usually encoded by a single-genome region called an S-locus, containing several multiallelic genes. The pollen and pistil are incompatible if both of them express identical alleles, otherwise they are compatible (de Nettancourt, 1977). In gametophytic SI systems, the phenotype of pollen is determined by the gametophyte (that is, by its own haploid genome), whereas in sporophytic SI systems, it is determined by the sporophyte (that is, by the diploid pollen parent) and there can be dominance interactions among alleles. As the SI systems constitute invisible barriers to self-fertilization and to mating between closely related individuals; the presence of the system probably has a large influence on actual reproduction patterns in a local deme. Recently, Schierup et al. (2006) studied mate availability and mating events by directly genotyping S-alleles between two consecutive generations in a natural deme of Arabidopsis lyrata (Brassicaceae). In Prunus avium populations, Stoeckel et al. (2011) also studied the effects of SI system and genetic drift by comparing the frequencies of S-alleles between multiple groups of parents and offspring using a mating model that included the S-locus data. These studies demonstrated that not only SI systems but also genetic drift, limited pollen dispersal, large reproductive variance and other factors affected mating patterns. However, direct investigations of the influence of SI systems on mating patterns in natural populations are scarce.
The evolutionary properties of SI systems have long aroused the interest of population geneticists studying balancing selection. As compatible crosses require distinct alleles, a direct expected consequence is that rare alleles are favored within populations, such that S-alleles are subject to negative frequency-dependent selection (NFDS) (for a review see Lawrence, 2000). Thus, a greater number of alleles are expected at the S-locus than at selectively neutral loci. Several theoretical studies have predicted that the patterns of polymorphism among populations are different between genes subject to NFDS and selectively neutral loci (Schierup, 1998; Schierup et al., 2000; Muirhead, 2001). NFDS that favors rare migrant alleles is expected to increase effective migration rates, so that the alleles tend to be shared among populations. Consequently, the degree of differentiation among populations is expected to be less (lower GST and FST values) at the S-locus than at selectively neutral loci, and thus unlike the selectively neutral alleles, the number of S-alleles within populations should be quite independent of population subdivision (Schierup, 1998; Schierup et al., 2000; Muirhead, 2001).
Several empirical tests for a coherent set of such predictions have been now undertaken in natural populations of several plant species. In Brassica insularis, Glémin et al. (2005) compared the population structure at the S-locus with that indicated by selectively neutral loci, nuclear simple sequence repeats (SSRs or microsatellites); their data supported most of the theoretical predictions. In Senecio squalidus, spatial genetic structure within populations was found to be weak at both the S-locus and at allozyme loci for which variation seemed to be selectively neutral (Brennan et al., 2003), whereas genetic differentiation among populations was smaller at the S-locus than at the allozyme loci (Brennan et al., 2006). In Arabidopsis halleri, the spatial genetic structure within populations was weaker when determined on the basis of the S-locus than when SSR loci were considered (Leducq et al., 2011). In P. avium, genetic differentiation among populations was smaller according to S-locus data than SSR loci (Stoeckel et al., 2008); higher diversity and evenness in allele frequencies, as well as larger deviations from neutrality at the S-locus than at the SSR loci were observed in a local population, but the spatial genetic structure within the population did not differ between these loci (Schueler et al., 2006). In Pyrus pyraster, genetic differentiation among populations was smaller according to S-locus data than SSR loci (Holderegger et al., 2008).
In this study, we evaluated both direct effects of SI systems on mating patterns in local demes and the effects of NFDS resulting from the mating process on population genetic structure. In order to address these aims, natural populations on islands were investigated. Island populations were chosen because observations of mating patterns are likely to be accurate in isolated demes on islands, and because infrequent migration between islands is likely to result in obvious genetic structure among island populations. P. lannesiana var. speciosa is suitable for this approach as it has a gametophytic SI system for which the molecular mechanism is already known, thus allowing us to genotype the S-locus directly in individuals sampled from natural populations (Kato and Mukai, 2004). In order to test the theoretical predictions for NFDS at the S-locus, SSRs were also examined to represent selectively neutral loci. First, the mating patterns in local demes were determined by paternity assignment, and the effects of SI system on mating success and changes in allele frequencies during the mating processes were evaluated. Second, the genetic structure within and among the populations was assessed at various spatial scales.
Materials and methods
Study species and sites
P. lannesiana var. speciosa (Rosaceae) is a deciduous tree species, semi-endemic to the Izu Islands, south of Honshu, Japan. Insects, such as bees, flies and beetles, pollinate the flowers and birds disperse the seeds (Kato, S., personal observation).
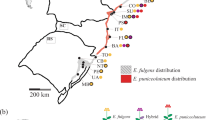
Study sites were selected from the whole range of the species’ distribution, consisting of the Izu Peninsula and seven islands: Oshima, Niijima, Kouzu, Miyake, Mikura, Hachijo and Aogashima (Figure 1a). A single site was selected on the peninsula and on each of the islands, with the exception of the Hachijo Island, where three subsites, HcA, HcB and HcC, were chosen (Figure 1b). Adult trees with a diameter at breast height (dbh)>10 cm were sampled from the seven sites and the three subsites to assess the genetic structure across the peninsula and island populations (trees from the three subsites on the Hachijo Island were pooled) (Figure 1b). In our previous studies, putative hybrids with related species were found on the Izu Peninsula (Kato et al., 2007, 2011). Such individuals were excluded from the samples collected from the Izu Peninsula site in this study.

Locations of populations and numbers of individuals of P. lannesiana var. speciosa examined in this study. Eight populations on the Izu Peninsula and Izu Islands (a), three sub-populations, HcA with a juvenile cohort, HcB, and HcC, and two plots, A and B on Hachijo Island (b), and locations and crown areas of trees in each plot (c). Trees with shaded crowns are selected as mother trees.
On the Hachijo Island, juvenile trees (<5 cm dbh) were sampled at subsite HcA (Figure 1b) to compare adult and juvenile cohorts, and two plots, A and B, were also established in order to observe mating patterns (Figure 1b). In each plot, all flowering trees were mapped, and seven mother trees were chosen (Figure 1c). The crown area (m2) of every flowering tree was measured (Figure 1c). Plot A was located in a forest, where the flowering trees occurred occasionally, and there were a few potential mates outside the plot. Plot B was in a meadow on the north-facing slope of the mountain and was quite isolated (>300 m away) from other flowering trees outside the plot.
Genotyping samples at the S-locus and SSR loci
Young leaves of adult trees at the eight sites, including the three subsites, juvenile trees from subsite HcA and all flowering trees in the two plots, were sampled. Seeds of the 14 mother trees in these plots were also collected in 2001 and/or 2002. The first leaves of seedlings developing from these seeds were sampled once the cotyledons appeared. As most (>90%) of the seeds germinated, differences in allele frequencies between the seeds and seedlings could be ignored. The sampled leaves were stored at –80 °C while awaiting DNA extraction.
Total DNA was extracted from the stored leaves using the method described by Murray and Thompson (1980). Genotypes at the S-locus, sequences of which were confirmed to be an S-RNase gene (Kato et al., 2007), were determined based on restriction fragment length polymorphism (RFLP) detected by genomic Southern analysis using complementary DNA for S-RNase as a probe, as described in Kato and Mukai (2004). This method could certainly visualize the variations of both two S-alleles, which respective individuals possess, implying that null alleles are unlikely to be detected at the S-locus. Genotypes at 11 nuclear SSR loci, BPPCT005, 014, 026, 028, 034, 037, 038 and 040 (Dirlewanger et al., 2002), UDP96-001 (Cipriani et al., 1999), UDP98-412 (Testolin et al., 2000) and pchcms5 (Sosinski et al., 2000), were also scored using the method described by Kato et al. (2011). Part of the genotypes had been already determined in our previous studies (Kato and Mukai, 2004; Kato et al., 2007, 2011).
Mating patterns
Paternity of every genotyped seed was assigned by means of simple exclusion based on genotypes at the S-locus and SSR loci of all flowering trees in each plot. If it was not possible to assign paternity, the pollen donor was assumed to be located outside the plot. When multiple pollen donors were assigned, the maximum-likelihood paternity was determined using Cervus ver. 2.0 (Marshall, 2001) with a 95% threshold as the strict confidence level. Allele frequencies of the flowering trees in each plot were used in an assignment simulation with the parameters: 10 000 cycles, 99% of loci typed and 0% error rate. For both S-locus and SSR loci, the genotyping was assumed to be accurate because no mismatch of genotypes was found between the seeds and their mother trees. Only flowering trees were regarded as candidate pollen donors in each plot. The proportions of candidate pollen donors sampled were 60% in plot A and 80% in plot B in consideration of the isolation from other flowering trees outside these plots.
Effects of SI on mating success
Effects of the SI system on male–female pair fecundity were evaluated using a Bayesian model (Klein et al., 2008, Tani et al., 2009), discriminating between factors such as spatial distance, kinship coefficient, flowering synchrony between mates and floral abundance of pollen donors. The SI score s between the mother trees and the candidate pollen donors was allocated a value of 0 when they were compatible (no S-alleles were shared), 0.5 when half-compatible (one of two alleles at the S-locus were shared) and 1 when incompatible (both S-alleles were shared). The horizontal distances, d (m), between the mother trees and the other flowering trees were recorded in each plot. The kinship coefficient g (Loiselle et al., 1995) between them was calculated for each plot using SPAGeDi version 1.1 (Hardy and Vekemans, 2002). In order to measure the flowering synchrony, the relative flowering intensity r of every flowering tree in each plot was scored every day from 2 March to 10 April in 2002 at six levels from 0 (no flowers were open or almost all the petals had fallen off), to 1, 2, 3 or 4 (1/5, 2/5, 3/5 and 4/5 of flowers, respectively, were opening or remaining) and 5 (all flowers were open). Flowering synchrony oij between the mother tree i and the candidate pollen donor j was defined as:

where t is the observation date. The floral abundance of the flowering trees was based on their crown area c (m2). The relative effects of individual factors (SI score, kinship coefficient, flowering synchrony and floral abundance) were evaluated by means of standardization of variables of the factors, which makes the variables to have mean=0 and s.d.=1. For the standardized variables, corresponding parameters were estimated.
Parameters of pollen dispersal and male fecundity have often been estimated on the basis of the neighborhood model using maximum likelihood (Burczyk et al., 2002) or Bayesian methods (Klein et al., 2008). In our mating model, the results of paternity assignment were used to simplify the model (Tani et al., 2009), and an exponential power function (Oddou-Muratorio et al., 2005) was applied to a dispersal kernel. Based on the paternity assignment, mother trees with >10 seeds, whose pollen donors were found within the plots, were examined in our model. For mother tree i {i=1, 2,..., M}, the number of seeds nij sired by pollen donor j {j=1, 2,..., N}, including selfing when i and j were the same tree, was assumed to follow a multinomial distribution with probability pij and sample size  . The probability pij that pollen donor j sired seeds of mother tree i was determined by parameters of pollen dispersal α1 (scaling), α2 (shape) and male–female pair fecundity fij from pollen donor j to mother tree i in the form:
. The probability pij that pollen donor j sired seeds of mother tree i was determined by parameters of pollen dispersal α1 (scaling), α2 (shape) and male–female pair fecundity fij from pollen donor j to mother tree i in the form:

where dij is the horizontal distance between mother tree i and pollen donor j. Male–female pair fecundity fij of pollen donor j to mother tree i was determined from SI score sij, kinship coefficient gij, flowering synchrony oij and crown area cj in the form:

where β1, β2, β3 and β4 are parameters of sij, gij, oij and cj, respectively. In 2001, β3 was not estimated because oij was not recorded. In each year, the parameters were estimated from both plots A and B, assuming that the same parameter was shared between the plots. The likelihood function for M mother trees was expressed as:

where mother trees and pollen donors were iA {iA=1, 2,..., MA} and jA {jA=1, 2,..., NA} in plot A and iB {iB=1, 2,..., MB} and jB {jB=1, 2,..., NB} in plot B, respectively. The posterior probability distributions were derived from the likelihood and prior probabilities of normal distributions with mean 0 and variance 1,000 for β1, β2, β3 and β4 (initial values were 0), and prior probabilities of gamma distributions with their parameters set to 0.001 for α1 and α2 (initial values were 1). The parameters were estimated as posterior medians via Markov-chain Monte Carlo (MCMC) sampling using JAGS 2.1.0 (Plummer, 2010) and the rjags package in R 2.11.1 (R Development Core Team 2010). The MCMC procedure for the model was run for 50 000 iterations with 100 intervals, after a burn-in period of 5,000 iterations. Convergence of the MCMCs was observed after all iterations of three chains of the parameters, and these were visualized using the coda package in R 2.11.1.
Allele frequencies at various stages
The allele frequencies in potential mates were obtained from the flowering trees in each plot. In order to obtain allele frequencies in the pollen that fertilized offspring of mother trees, a paternal gametic genotype at each locus for each offspring was determined by subtracting the maternal gametic genotype from the offspring genotype. It was possible to determine the paternal gamete genotype unambiguously unless both an offspring and its parents were heterozygous with the same pair of alleles (for example, genotype AB). Otherwise, the paternal gamete genotype was either A or B at a frequency of 0.5.
Changes in allele frequencies were compared between the potential mates and pollen siring offspring of the mother trees in each plot. Between these two generations, the NFDS effect caused by the SI system is expected to lead to the changes in allele frequencies at the S-locus. In a deterministic model, alleles at the S-locus with higher and lower frequencies in potential mates will decrease and increase in siring pollen, respectively. However, such changes at the SSR loci are not expected. When the changes calculated by subtracting the allele frequencies at the S-locus and SSR loci in the potential mates from those in the siring pollen are plotted against the allele frequencies in the potential mates in each plot, a negative correlation is expected only at the loci under NFDS, that is, the S-locus.
In addition to the NFDS effect, genetic drift results in variations in allele frequencies due to the finite size of sampled offspring through a random sampling process. The variations were generated from Monte Carlo simulations for both the S-locus and SSR loci because random genetic drift affects both types of loci. The number of alleles was assumed to follow a multinomial distribution with the sample size (the number of offspring sampled from the mother trees in each plot) and the allele frequencies in the potential mates in each plot. The procedure was repeated 1,000 times, and the 95% confidence interval for each allele frequency at each locus in the potential mates was obtained. From the confidence interval, sampling variances in the changes in allele frequencies from siring pollen to potential mates were also plotted to evaluate the effect of random genetic drift.
Between the adult and juvenile cohorts at subsite HcA, the changes in allele frequencies were also examined by the same method because NFDS is also expected to occur during the establishment of the juvenile cohort from the adult cohort through reproduction and regeneration.
Linkage disequilibria and neutrality
The linkage among the S-locus and the SSR loci and their selective neutrality were examined. The linkage disequilibrium at every pair of these loci within and across adult populations at the eight sites was tested after sequential Bonferroni multiple-comparison correction using FSTAT 2.9.3.2 (Goudet, 2001). The selective neutrality at the S-locus and the SSR loci in the eight adult populations was evaluated by comparing observed and expected allele frequencies, according to Ewens’ sampling theory (Ewens, 1972) and using Watterson’s homozygosity test (Watterson, 1978) implemented in Arlequin 2.0 (Schneider et al., 2000).
Spatial genetic structure
Spatial genetic structure was compared between the S-locus and the SSR loci at various scales. At the local scale (within the plots), spatial autocorrelation was assessed by means of the kinship coefficient gij, which measures the correlation between the frequencies of homologous alleles in pairs of individuals (Loiselle et al., 1995). Multiallelic single-locus and the multilocus kinship estimators were plotted against the logarithm of geographical distance, and the linear regression slope bg was tested for significance by Mantel tests with 10 000 permutations. For the SSR loci, the mean and s.e. of bg were estimated using a jackknife procedure over loci. In order to evaluate whether this mean is significantly larger than the value of bg at the S-locus, we compared using a one-tailed t-test. The standardized regression slope (Sp) statistic, defined as Sp=–bg/(1–g1) in Vekemans and Hardy (2004), was also calculated. Here g1 is the kinship estimator gij between adjacent individuals. The Sp statistic allows a better comparison between different species and populations, as the kinship estimator itself depends on the sampling scheme used (Vekemans and Hardy, 2004). Calculations of the pairwise gij, the Mantel tests and the jack-knife tests over loci were performed using SPAGeDi.
At the regional scale (among the eight sites), isolation-by-distance was examined by means of the relationship between the genetic distance between the adult populations and the log-transformed geographic distance between the sites (Rousset, 1997). The pairwise population GST values were calculated as described below, and used as the genetic distance indices. The significance of the association between the two types of distance was determined by the Mantel test (Mantel, 1967) using permutation procedures (1000 resamplings) implemented in the ISOLDE module of GENEPOP 4.0.10 (Raymond and Rousset, 1995).
Population genetic structure
The total and effective numbers of alleles (Kimura and Crow, 1964) and the observed and expected heterozygosity (Nei, 1973) across the eight adult populations were also calculated for the S-locus and the SSR loci. Genetic differentiation among the eight sites and among the three subsites was evaluated using a standard measure, GST value. The 95% confidence interval and the P value for GST were obtained by bootstrapping using the DEMEtics package in R 2.11.1.
Muirhead (2001) suggested that GST and FST are not the best statistics for revealing population differentiation under balancing selection and that the pattern of allele sharing among populations (sharing distribution) is better. The sharing distribution is a frequency distribution of alleles shared among k populations (k=1, 2, …, n) in a total of n populations. We determined this distribution for both the S-locus and SSR loci from adult populations at the eight sites.
Results
In total, 439 adult trees were sampled from the Izu peninsula and the seven islands, and the sample size ranged from 31 to 98 among the eight sites (Figure 1a). The 98 adult trees on the Hachijo Island were divided into the three subsites, HcA, HcB and HcC, with sample sizes of 33, 33 and 32, respectively (Figure 1b). At subsite HcA, 26 juvenile trees were also sampled (Figure 1b). The average dbhs of the adult and juvenile trees at subsite HcA were 21.5±1.3 (mean±s.e.) and 2.7±0.2 cm, respectively. On the Hachijo Island, 86 flowering trees in plot A and 79 in plot B were examined as candidate pollen donors (Figure 1c). The average crown area of the flowering trees was 11.1±1.0 (mean±s.e.) m2 in plot A and 6.8±0.6 m2 in plot B (Figure 1c).
Mating patterns and effects of SI on mating success
Two monomorphic SSR loci, BPPCT026 and UDP96-001, found in the two plots were not used in the following paternity assignment. The exclusion probability for paternity over the remaining nine SSR loci and the S-locus was 0.998 in plot A and 0.997 in plot B. The pollen donors for 88 out of 152 (57.9%) seeds of seven mother trees in plot A and 179 out of 221 (81.0%) seeds of seven mother trees in plot B were assigned within the plots (Table 1). A single seed in plot A was the result of self-pollination. The mean distances from the mother trees to the assigned pollen donors for their seeds ranged from 8.4 to 19.3 m in plot A and from 9.9 to 22.0 m in plot B (Table 1).
The parameters estimated from the mating models for pollen dispersal and male–female pair fecundity was similar in 2001 and 2002 (Table 2). According to the estimated scale (α1) and shape (α2) parameters for pollen dispersal, mean dispersal distance α1Γ(3/α2)/Γ(2/α2) (Oddou-Muratorio et al., 2005) was 6.7 m in 2001 and 13.5 m in 2002, and the dispersal curves were more leptokurtic (α2=0.57 in 2001 and 0.52 in 2002) than an exponential curve (α2=1). With respect to male–female pair fecundity, the SI score and the kinship coefficient had significantly negative effects, whereas flowering synchrony and floral abundance had significantly positive effects because their 95% credible intervals did not include 0 (Table 2). The floral abundance had the largest effect among the four factors (Table 2). Male–female pair fecundity decreased not only in incompatible mating pairs, but also in half-compatible pairs, in comparison with fully compatible pairs (Figures 2a and c). The kinship coefficient showed negative associations with male–female pair fecundity in overall patterns, but indicated a non-linear relationship, where the fecundity decreased, as a result of both positive and negative kinship coefficients, that is, with increasing deviation from 0 (Figures 2b and d).

Relationships of male–female pair fecundity with the incompatibility index based on the S-locus data in 2001 (a) and 2002 (c) and with the kinship coefficient calculated from the SSR loci data in 2001 (b) and 2002 (d). The data for nine mother trees in plots A and B were pooled.
Allele frequencies
The changes in allele frequencies at the S-locus from potential mates to siring pollen showed a significant negative correlation with allele frequencies in the potential mates in plot B (R=–0.464, P=0.030) although no significant correlation was found in plot A (R=–0.383, P=0.078; Figures 3a and c). In the SSR loci, no significant correlation was found in the both plots (R=–0.245, P=0.068 in plot A and R=–0.149, P=0.274 in plot B; Figures 3b and d). The changes in allele frequencies from adult to juvenile cohorts were not significantly correlated with allele frequencies in the adult cohorts at both the S-locus and SSR loci (R=–0.280, P=0.218 for S-locus and R=0.040, P=0.752 for SSR loci; Figures 3e and f). At both the S-locus and SSR loci, some alleles showed significant changes in allele frequencies, which exceeded the 95% confidence intervals of sampling variances due to random genetic drift (Figure 3).

Correlations between change in allele frequencies from potential mates to siring pollen and allele frequencies in the potential mates at the S-locus in plots A (a) and B (c) and at the SSR loci in plots A (b) and B (d) and between change in allele frequencies from adult to juvenile cohorts and allele frequencies in the adult cohorts at the S-locus (e) and SSR loci (f). Dotted lines show the 95% confidence intervals of sampling variances due to random genetic drift.
Linkage disequilibria and neutrality
No pairings of the S-locus and the SSR loci showed significant linkage disequilibrium across the eight adult populations or within the populations individually (P>adjusted P=0.00076).
Significant deviations from selective neutrality were detected at the S-locus in adult populations at seven of the eight sites; the exception was Aogashima (Watterson’s homozygosity test, P<0.05). Among the SSR loci, significant deviations from neutrality were found at BPPCT034 and 038 at a single site (P<0.05) and at BPPCT014 and 028 at two sites (P<0.05).
Spatial genetic structure
At the local scale, significant decreases in pairwise kinship coefficients with increasing spatial distance in both the plots were detected at the SSR loci (Mantel test, P<0.05; Table 3), but not at the S-locus. Among the nine SSR loci that were not monomorphic, that is, excluding BPPCT026 and UDP96-001, there was considerable variance in the slope bg. In plot A, the bg value for the S-locus were significantly smaller (an almost sixfold decrease) than those for the SSR loci (one-tailed t-test, P<0.05), but it was not the case in plot B. The Sp values at the S-locus (0.00139 in plot A and 0.00450 in plot B) were smaller than the multilocus Sp values at the SSR loci (0.00807 in plot A and 0.00729 in plot B).
At the regional scale, significant decreases in the pairwise measures of genetic differentiation (GST value) with increasing geographical distance were found at the S-locus and at nine SSR loci, the exceptions being BPPCT005 and 034 (Mantel test, P<0.05; Table 3). The regression slope (b) was smaller at the S-locus (0.012) than at the SSR loci (0.039) (Table 3).
Population genetic structure
Across adult populations at the eight sites, both the total number and effective number of alleles at the S-locus were much higher than at the SSR loci (Table 4). The expected heterozygosity was also higher at the S-locus than at the SSR loci (Table 4), although obligate heterozygosity was not found at the S-locus because there was a single homozygous adult tree.
Among adult populations at the three subsites on the Hachijo Island, genetic differentiation measured by GST value was significant at both the S-locus and the three SSR loci (P<0.05; Table 4). The GST value at the S-locus (0.005) was lower than the mean GST value over the SSR loci (0.011) although their 95% confidence intervals overlapped (Table 4). Among adult populations at the eight sites, GST value was significantly positive at both the S-locus and the 11 SSR loci (P<0.001; Table 4). The GST value at the S-locus (0.020) was significantly lower than the mean GST value over the SSR loci (0.065) because their 95% confidence intervals did not overlap (Table 4).
For the S-locus and the SSR loci, frequency distributions of alleles shared among k populations (k=1, 2, …, 8) (sharing distributions) were determined for adult populations at the eight sites. Alleles found only in a single population (private alleles) were the most frequent alleles at both the S-locus and SSR loci (Figure 4). Excluding the private alleles, the sharing distribution for the S-locus was bell-shaped with a peak at alleles shared among five populations (Figure 4a). In contrast, the sharing distribution for the mean frequency across the SSR loci was U-shaped, with two peaks, one associated with the private alleles and the other with the alleles common to all the populations (Figure 4b).

Distributions of allele-sharing probabilities of the S-locus (a) and SSR loci (b) in eight adult populations.
Discussion
The S-locus and SSR loci examined in this study are regarded as unlinked, independent loci because no linkage disequilibrium was detected in any pairs of the loci. Allelic diversity was clearly higher at the S-locus than at the SSR loci (Table 4). The result indicates the NFDS effect at the S-locus, assuming that the SSR loci are selectively neutral. The occurrence of NFDS at the S-locus and the approximate neutrality at the SSR loci were supported by the neutrality tests. These tests revealed significant departures from neutrality at the S-locus in seven of the eight populations, while they detected significant deviations at four of the eleven SSR loci in one or two populations.
Effects of the gametophytic SI system on mating patterns
In our samples, one seed and one tree were considered to result from self-pollination. This result is unlikely to indicate the presence of a self-compatible allele because the behavior of the S-alleles of the seed and the tree fulfill a normal function of the SI system in the other seeds and trees. As our analytical method is less likely to lead to genotyping errors, the putative selfed samples may imply pseudo-self-compatibility that rarely occurs by chance.
We evaluated the influence of a gametophytic SI system on mating patterns in two local demes (plots A and B) of P. lannesiana var. speciosa. As the S-alleles exhibited high diversity in the plots (Kato and Mukai, 2004), almost all the pairs of mother trees and candidate pollen donors were either fully compatible (67.9%) or half-compatible (29.9%). Therefore, the negative effect of the SI scores on mating success was mainly a result of differences in the scores between fully compatible (1) and half-compatible (0.5) pairs (Figures 2a and c). In the half-compatible pairs, half of the pollen from the donors can pollinate the mother trees as a result of the gametophytic SI system. Pollen deposited on a stigma competes for access to the ovules, each of which develops into a single seed in P. lannesiana var. speciosa. If pollen from half and fully compatible donors is mixed on a stigma, the half-compatible donor will be half as likely to sire offspring as the fully compatible donor as a result of pollen competition. On the other hand, if each flower only receives pollen from either a half or a fully compatible donor because of limited mating opportunity, the half-compatible donor will be equally likely to sire offspring as the fully compatible donor. Thus, the recorded reduction in siring success of half-compatible donors suggests that there was mixed pollination, with pollen reaching the flowers from both half and fully compatible donors, with sufficient availability of multiple mates. In line with our results, pollen donor diversity was high in a fragmented P. avium population (Cottrell et al., 2009), and multiple mates were available in an isolated P. mahaleb population (Garcíia et al., 2005). Holderegger et al. (2008) concluded that populations of rare wild pear (Pyrus pyraster) were not limited in terms of principal mate availability even if the S-allele diversity was smaller. The wild pear also has a gametophytic SI system, and thus both half and fully compatible pairs occur in their mating. As Holderegger et al. (2008) did not discriminate half and fully compatible pairs, the difference in fecundity between the half and fully compatible pairs shown in the study had not been demonstrated in their study.
The realized mating patterns in natural conditions may depend not only on the SI system but also on pollen dispersal and fecundity variations in local demes. The pollen dispersal distances estimated from the mating model were 6.7 and 13.5 m, and the actual dispersal distances ranged from 8.4 to 22.0 m (Table 1). These distances were shorter than those recorded for some animal-pollinated plants (Smouse and Sork, 2004) and Prunus populations (Garcíia et al., 2005; Cottrell et al., 2009), although there was some long distance pollination from outside of the plots. Schierup et al. (2006) reported that pollen donors siring seeds appeared to occur closer to the mother plants than compatible candidates in a population of Arabidopsis lyrata. Cottrell et al. (2009) also found a similar pattern in a population of P. avium. Also in P. avium, Stoeckel et al. (2011) suggested that the effect of local drift associated with limited pollen dispersal and large reproductive variance was strong enough to counteract the influence of SI systems on mating patterns. Our findings also suggest that available mates are limited to neighbors, resulting in the spatial genetic structure observed in the local demes (Table 3). Male–female pair fecundity mainly depended on the crown area of pollen donors, suggesting that the amount of pollen produced most greatly affects mating success (Table 2). Flowering synchrony also restricted mating pairs, as indicated by its significant positive effect on mating success. Furthermore, mating between close relatives can increase the risk of selective abortion and inbreeding depression (Charlesworth and Charlesworth, 1987; Charlesworth et al., 1990), which is consistent with the overall negative effects of the kinship coefficient on mating success. The kinship coefficient tended to have more complicated effects than the SI scores. Male–female pair fecundity decreased when relatedness was either positive or negative (Figures 2b and d), suggesting that mating between distant relatives is also prevented, although it is difficult to determine the exact causes. The lower fecundity in pairs with low kinship could be due to outbreeding depression although outbreeding depression was not detected in P. mahaleb using crossing experiments (Pflugshaupt et al., 2002).
As mating under a gametophytic SI system requires distinct S-alleles between the male and female, the direct expected consequence is that rare S-alleles frequencies increase in the next generation whereas very common S-alleles are counter-selected. In P. avium populations, Stoeckel et al. (2011) firstly provided the formal empirical demonstration of this direct NFDS effect. Our observation in plot B also revealed the changes in allele frequencies at the S-locus from potential mates to mating pollen, as expected under NFDS, by comparing between the S-locus and SSR loci (Figure 3). However, we failed to find the change in plot A, where the proportion of seeds whose pollen donors were assigned within the plots was lower (58%) than in plot B (81%). Thus, lower reliability of allele frequency in the potential mates in plot A may lead to the result. However, between the two cohorts of juvenile and adult trees, such changes were not apparent at any loci, indicating that the survival process after the fertilization makes allele frequency patterns produced by the SI system too noisy to detect the NFDS effect.
Our findings suggest that the gametophytic SI system with half and full compatibility affects mating patterns in the local demes, as well as preventing self-fertilization in P. lannesiana var. speciosa. Various ecological and genetic factors, such as limited pollen dispersal, floral abundance, flowering synchrony, and kinship, are also likely to affect the mating patterns. The NFDS effect owing to the SI system is evident although mating biases due to the ecological and genetic factors and the stochastic regeneration process could mask the expected changes in allele frequencies at the S-locus.
Population structure at the S-locus
Theoretical predictions for balancing selection are lower genetic differentiation between populations and less-clear spatial genetic structure at the S-locus in comparison with that at selectively neutral9 loci (Schierup, 1998; Schierup et al., 2000; Muirhead, 2001; Leducq et al., 2011). Glémin et al., 2005 compared genetic structure between the S-locus and SSR loci in five populations of Brassica insularis with a sporophytic SI system, and their results were consistent with these predictions. Although Brennan et al., 2003 found no significant spatial genetic structure at either the S-locus or allozyme loci in Senecio aqualidus, Leducq et al. (2011) recorded reduced spatial genetic structure at the S-locus than at SSR loci in Arabidopsis halleri. In rosaceous species with a gametophytic SI system, Stoeckel et al. (2008) and Holderegger et al. (2008) found less genetic differentiation between populations at the S-locus than at SSR loci, although Schueler et al. (2006) did not find any differences in the extent of spatial genetic structure between the two locus types. Our results showed less genetic differentiation between populations and reduced spatial genetic structure, as indicated by the GST values and kinship coefficients, at the S-locus than at the SSR loci at different spatial scales (Tables 3 and 4). Unlike the genetic differentiations among populations, the signature of NFDS effects on spatial genetic structure is expected to depend strongly on population and species characteristics, such as the extent of pollen and seed dispersal, the degree of population isolation, the type of SI system and the allelic diversity at the S-locus (Leducq et al., 2011). It is predicted that, for a certain allelic diversity at the S-locus, limited gene dispersal in isolated populations tends to increase difference in the NFDS effects on spatial genetic structure between the S-locus and SSR loci. Although the relatively narrow areas of the plots were investigated in our study, the gene dispersal within plots and the pollen immigration from outside the plots were restricted in comparison with those recorded from the other related species (P. avium in Schueler et al., 2006 and Cottrell et al., 2009, P. mahaleb in Garcíia et al., 2005). Thus, less-clear spatial genetic structure at the S-locus observed in P. lannesiana populations was consistent with the expectations in Leducq et al. (2011).
The sharing distribution at the S-locus observed in P. lannesiana populations was characterized by both a predominant occurrence of alleles unique to a single population (private alleles) and a bell-shaped distribution with a peak at alleles shared among five populations (Figure 4). This pattern is different from that observed in B. insularis populations, which are characterized by both the most frequent private alleles and the frequency decreasing as the number of populations increases (Glémin et al., 2005). Although the sharing distribution in B. insularis populations suggests a constantly low migration rate, that in P. lannesiana populations implies that the migration rate varies both spatially and temporally. The Izu Islands are oceanic islands without any previous connections to the mainland or between each other, with the exception of the Niijima and Kouzu Islands, which may have been joined at times of the minimum sea levels during glacial periods (Kaneoka et al., 1970). Genetic structure in P. lannesiana var. speciosa was characterized by relatively low genetic variation among island populations and isolation-by-distance patterns at both chloroplast and nuclear loci, suggesting seed-mediated gene flow by birds between neighboring islands (Kato et al., 2011). The genetic variation at the S-locus also exhibited an isolation-by-distance pattern (Kato et al., 2007). Thus, gene flow between islands that are close together and isolation of remote islands seem to be responsible for the allele sharing among populations and the accumulation of private alleles, respectively, at the S-locus. Furthermore, the Izu Islands were formed by volcanic activity a few million years ago (Kaneoka et al., 1970) and have suffered, historically, from volcanic eruptions (Kato et al., 2011). Such island history suggests colonization after the creation of new habitats and subsequent isolation of the established populations, which may explain the observed allele-sharing pattern at the S-locus. In contrast to the S-locus, the SSR loci exhibited a U-shaped sharing distribution with two peaks, one associated with the private alleles and the other with alleles common to all populations (Figure 4). Such U-shaped patterns were generated by positive selection and selective neutrality (Slatkin, 1980), while bell-shaped patterns are caused by balancing selection (Muirhead, 2001). Ignoring the most frequent private alleles, which are probably the consequences of recent isolation between islands, NFDS at the S-alleles in P. lannesiana var. speciosa is likely to have been responsible for allele-sharing patterns among the island populations.
Data archiving
Data have been deposited at Dryad: doi:10.5061/dryad.7c425.
References
Brennan AC, Harris SA, Hiscock SJ (2003). The population genetics of sporophytic self-incompatibility in Senecio squalidus L. (Asteraceae) II: a spatial autocorrelation approach to determining mating behaviour in the presence of low S allele diversity. Heredity 91: 502–509.
Brennan AC, Harris SA, Hiscock SJ (2006). The population genetics of sporophytic self-incompatibility in Senecio Squalidus L. (Asteraceae): the number, frequency, and dominance interactions of S alleles across its British range. Evolution 60: 213–224.
Burczyk J, Adams WT, Moran GF, Griffin AR (2002). Complex patterns of mating revealed in a Eucalyptus regnans seed orchard using allozyme markers and the neighbourhood model. Mol Ecol 11: 2379–2391.
Charlesworth D, Charlesworth B (1987). Inbreeding depression and its evolutionary consequences. Annu Rev Ecol Systematics 18: 237–268.
Charlesworth D, Morgan MT, Charlesworth B (1990). Inbreeding depression, genetic load, and the evolution of outcrossing rates in multilocus system with no linkage. Evolution 44: 1469–1489.
Cipriani G, Lot G, Huang WG, Marrazzo MT, Peterlunger E, Testolin R (1999). AC/GT and AG/CT microsatellite repeats in peach [Prunus persica (L) Batsch]: isolation, characterisation and cross-species amplification in Prunus. Theor Appl Genet 99: 65–72.
Cottrell JE, Vaughan SP, Connolly T, Sing L, Moodley DJ, Russell K (2009). Contemporary pollen flow, characterization of the maternal ecological neighbourhood and mating patterns in wild cherry (Prunus avium L.). Heredity 103: 118–128.
de Nettancourt D (1977) Incompatibility in angiosperms. Springer-Verlag Berlin.
Dirlewanger E, Cosson P, Tavaud M, Aranzana MJ, Poizat C, Zanetto A et al (2002). Development of microsatellite markers in peach [Prunus persica (L.) Batsch] and their use in genetic diversity analysis in peach and sweet cherry (Prunus avium L.). Theor Appl Genet 105: 127–138.
Ewens WJ (1972). The sampling theory of selectively neutral alleles. Theor Popul Biol 3: 87–112.
Garcíia C, Arroy JM, Godoy JA, Jordano P (2005). Mating patterns, pollen dispersal, and the ecological maternal neighbourhood in a Prunus mahaleb L. population. Mol Ecol 14: 1821–1830.
Glémin S, Gaude T, Guillemin ML, Lourmas M, Olivieri I, Mignot A (2005). Balancing selection in the wild: testing population genetics theory of self-incompatibility in the rare species Brassica insularis. Genetics 171: 279–289.
Goudet J (2001) FSTAT; a program to estimate and test gene diversities and fixation indices version 2.9.3 http://www.unil.ch/izea/softwares/fstat.html.
Hardy OJ, Vekemans X (2002). SPAGEDI a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620.
Holderegger R, Häner R, Csencsics D, Angelone S, Hoebee SE (2008). S-allele diversity suggests no mate limitation in small populations of a self-incompatible plant. Evolution 62: 2922–2928.
Kaneoka I, Isshiki N, Zashu S (1970). K-Ar ages of the Izu-Bonin Islands. Geochem J 4: 53–60.
Kato S, Iwata H, Tsumura Y, Mukai Y (2007). Distribution of S-alleles in island populations of flowering cherry, Prunus lannesiana var. speciosa. Genes Genet Syst 82: 65–75.
Kato S, Iwata H, Tsumura Y, Mukai Y (2011). Genetic structure of island populations of Prunus lannesiana var. speciosa revealed by chloroplast DNA, AFLP and nuclear SSR loci analyses. J Plant Res 124: 11–23.
Kato S, Mukai Y (2004). Allelic diversity of S-RNase at the self-incompatibility locus in natural flowering cherry populations (Prunus lannesiana var. speciosa). Heredity 92: 249–256.
Kimura M, Crow JF (1964). The number of alleles that can be maintained in a finite population. Genetics 49: 725–738.
Klein EK, Desassis N, Oddou-Muratorio S (2008). Pollen flow in the wild service tree, Sorbus torminalis (L.) Crantz. IV. Whole interindividual variance of male fecundity estimated. Mol Ecol 17: 3323–3336.
Lawrence MJ (2000). Population genetics of the homomorphic self-incompatibility polymorphisms in flowering plants. Ann Bot 85: 221–226.
Leducq JB, Llaurens V, Castric V, Saumitou-Laprade P, Hardy OJ, Vekemans X (2011). Effect of balancing selection on spatial genetic structure within populations: theoretical investigations on the self-incompatibility locus and empirical studies in Arabidopsis halleri. Heredity 106: 319–329.
Loiselle BA, Sork VL, Nason J, Graham C (1995). Spatial genetic-structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am J Bot 82: 1420–1425.
Mantel NA (1967). The detection of disease clustering and a generalized regression approach. Cancer Res 27: 209–220.
Marshall TC (2001) CERVUS ver.2.0 Available at http://helios.bto.ed.ac.uk/evolgen.
Marshall TC, Slate J, Kruuk LEB, Pemberton JM (1998). Statistical confidence for likelihood-based paternity inference in natural populations. Mol Ecol 7: 639–655.
Muirhead CA (2001). Consequences of population structure on genes under balancing selection. Evolution 55: 1532–1541.
Murray MG, Thompson WF (1980). Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8: 4321–4325.
Nei M (1973). Analysis of gene diversity in subdivided populations. Proc Nat Acad Sci USA 70: 3321–3323.
Oddou-Muratorio S, Klein EK, Austerlitz F (2005). Pollen flow in the wild service tree, Sorbus torminalis (L.) Crantz. II. Pollen dispersal and heterogeneity in mating success inferred from parent-offspring analysis. Mol Ecol 14: 4441–4452.
Pflugshaupt K, Kollmann J, Fischer M, Roy B (2002). Pollen quantity and quality affect fruit abortion in small populations of a rare fleshy-fruited shrub. Basic Appl Ecol 3: 319–327.
Plummer M (2010) http://www-fis.iarc.fr/∼martyn/software/jags/.
Raymond M, Rousset F (1995). GENEPOP: population genetics software for exact test and ecumenicism. J Hered 86: 248–249.
Rousset F (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Schierup MH (1998). The number of self-incompatibility alleles in a finite, subdivided population. Genetics 149: 1153–1162.
Schierup MH, Bechsgaard JS, Nielsen LH, Christiansen FB (2006). Selection at work in self-incompatible Arabidopsis lyrata: mating patterns in a natural population. Genetics 172: 477–484.
Schierup MH, Vekemans X, Charlesworth D (2000). The effect of subdivision on variation at multi-allelic loci under balancing selection. Genet Res 76: 51–62.
Schneider S, Roessli D, Excoffier L (2000) ARLEQUIN, version 2.000: A Software for Population Genetics Data Analysis. Genetics and Biometry Laboratory, Department of Anthropology, University of Geneva Geneva, Switzerland.
Schueler S, Tusch A, Scholz F (2006). Comparative analysis of the within-population genetic structure in wild cherry (Prunus avium L.) at the self-incompatibility locus and nuclear microsatellites. Mol Ecol 15: 3231–3243.
Slatkin M (1980). The distribution of mutant alleles in a subdivided population. Genetics 95: 503–524.
Smouse PE, Sork VL (2004). Measuring pollen flow in forest trees: an exposition of alternative approaches. Forest Ecol Manag 197: 21–38.
Sosinski B, Gannavarapu M, Hager LD, Beck LE, King GJ, Ryder CD et al (2000). Characterization of microsatellite markers in peach Prunus persica (L.) Batsch. Theor Appl Genet 101: 421–428.
Stoeckel S, Castric V, Mariette S, Vekemans X (2008). Unequal allelic frequencies at the self-incompatibility locus within local populations of Prunus avium L.: an effect of population structure? J Evol Biol 21: 889–899.
Stoeckel S, Klein EK, Oddou-Muratorio S, Musch B, Mariette S (2011). Microevolution of S-allele frequencies in wild cherry populations: respective impacts of negative frequency dependent delection and genetic drift. Evolution 66: 486–504.
Tani N, Tsumura Y, Kado T, Taguchi Y, Lee SL, Muhammad N et al (2009). Paternity analysis-based inference of pollen dispersal patterns, male fecundity variation and influence of flowering tree density and general flowering magnitude in two dipterocarp species. Ann Bot 104: 1421–1434.
Testolin R, Marrazzo T, Cipriani G, Quarta R, Verde I, Dettori MT et al (2000). Microsatellite DNA in peach (Prunus percica L. Batsch) and its use fingerprinting and testing the genetic origin of cultivars. Genome 43: 512–520.
Vekemans X, Hardy OJ (2004). New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol 13: 921–935.
Watterson GA (1978). The homozygosity test of neutrality. Genetics 88: 405–417.
Acknowledgements
We thank Yoshinari Moriguchi and Tomokazu Takahashi for their practical with the sample collection, and Jungo Yuzurihara and Yasuomi Ota for their help in preserving the samples. We also thank Naoki Tani for providing his program code, which was used to estimate the parameters for pollen dispersal and male–female pair fecundity. This study was partly supported within the research budget of the Forest Recovery Project for the Disaster Area on Miyake Island, provided by the Tokyo Metropolitan Government, as well as a grant for Research on genetic guidelines based on molecular population analysis of plants for restoration projects from the Ministry of Environment, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Shuri, K., Saika, K., Junko, K. et al. Impact of negative frequency-dependent selection on mating pattern and genetic structure: a comparative analysis of the S-locus and nuclear SSR loci in Prunus lannesiana var. speciosa. Heredity 109, 188–198 (2012). https://doi.org/10.1038/hdy.2012.29
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2012.29
