
Abstract
The production of genome sequences has led to another important advance in their annotation, which is closely linked to the exact determination of their content in terms of repeats, among which are transposable elements (TEs). The evolutionary implications and the presence of coding regions in some TEs can confuse gene annotation, and also hinder the process of genome assembly, making particularly crucial to be able to annotate and classify them correctly in genome sequences. This review is intended to provide an overview as comprehensive as possible of the automated methods currently used to annotate and classify TEs in sequenced genomes. Different categories of programs exist according to their methodology and the repeat, which they can identify. I describe here the main characteristics of the programs, their main goals and the difficulties they can entail. The drawbacks of the different methods are also highlighted to help biologists who are unfamiliar with algorithmic methods to understand this methodology better. Globally, using several different programs and carrying out a cross comparison of their results has the best chance of finding reliable results as any single program. However, this makes it essential to verify the results provided by each program independently. The ideal solution would be to test all programs against the same data set to obtain a true comparison of their actual performance.
Similar content being viewed by others
Introduction
Over the past decade, genome sequencing has speeded up with the improvement of the various techniques used. The elucidation of genome sequences has made it obvious that one important step in the deciphering of these sequences involves the accurate determination of their repeat content. Repeats, and more particularly transposable elements (TEs), were initially considered to constitute only a negligible part of eukaryotic genomes, although long before sequencing began, it was known that these elements can sometimes account for a major proportion of genomes (Britten and Kohne, 1968). We now know that, depending on the organism, the proportion of TEs in the genome can differ widely, ranging from a few percent (3% in the yeast Saccharomyces cerevisiae; Kim et al., 1998) to a huge proportion encompassing almost the entire genome (>80% in the maize; SanMiguel et al., 1998), the human genome itself being particularly rich in repeats (which make up about 45%) (The International Human Genome Sequencing Consortium, 2001).
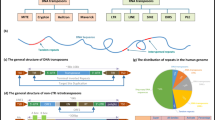
Repeats in genomes are classified on the basis of their sequence characteristics and of how they are formed. One category consists of tandem repeats, and includes any sequences found in consecutive copies along a DNA strand. Several different categories of tandem repeats have been defined, depending on the number of repeats and on the size of the repeated units. This group includes microsatellites or simple sequence repeats (short repeat units consisting of 1–6 nucleotides) and minisatellites (longer repeat units consisting of 10–60 nucleotides). Another category, on which this review will mainly focus, is constituted by elements that are found dispersed across the whole genome, and which consists mainly of TEs. TEs can be classified according to the intermediate they use to move (Finnegan, 1989). Class-I TEs use an RNA intermediate to transpose by a ‘copy and paste’ mechanism, whereas class-II TEs use a DNA intermediate, to transpose by a ‘cut and paste’ mechanism. Within each of these classes, TEs are further subdivided on the basis of the structural features of their sequences. The long terminal repeat (LTR) retrotransposons are class-I repeats with direct repeats called LTRs at their extremities and have coding capacities (Figure 1). This class of elements is distinguished from that of the non-LTR retrotransposons, which consists of two main subclasses: the long interspersed nuclear elements (LINEs), which have coding capacities, and the short interspersed nuclear elements (SINEs), which do not (Figure 1). The class-II TEs consist of the DNA transposons (Figure 1). More recently, it has been proposed that miniature inverted repeat transposable elements (MITEs), DNA-based elements but which move through a ‘copy and paste’ mechanism, could represent a new subclass of class-II elements (Wicker et al., 2007). The capacity of moving enables a given element to replicate itself, thus giving rise to a family that is represented by several copies of the same element. Given the relationship between elements, some families are phylogenetically related, which makes it possible to construct the evolutionary history of these elements (Capy et al., 1997).

The different types of TEs. The LTR retrotransposons have a primer binding site (PBS) on the 3’ side, and a polypurine tract (PPT) on the 5’ size. Some also present a third ORF coding for env. The pol gene is formed by different domains coding for a protease (PR), an integrase (INT), a reverse transcriptase (RT) and an RnaseH (RH), respectively. The non-LTR retrotransposons have a poly A tail at their 3’ extremity. The LINEs display two ORFs whereas the SINEs have no coding capacity. The autonomous helitrons possess a coding capacity for a helicase and an RPA-like (RAPl) single-stranded DNA-binding protein.
In addition to their numerical importance in genomes, which is what makes these elements responsible for the increase in genome size in most species, TEs are now known to have a major part in genome evolution (Biémont and Vieira, 2006). Their role includes genome rearrangement because of homologous recombination among copies of a given family, gene innovation through various mechanisms, such as exon shuffling, gene regulation through their own promoter regions, and insertional mutations by direct insertion into genes (Kidwell and Lisch, 2001). These various evolutionary implications and the presence of coding regions in some TEs can lead to confusion in gene annotation, and can also complicate the process of genome assembly (Tang, 2007), which makes it particularly crucial to be able to annotate and classify TEs correctly in genome sequences.
The problem of identifying repeats in sequences is a recurrent difficulty in algorithmics and the automated detection of such elements is no trivial task. It is particularly difficult to determine the real boundaries of these sequences accurately. They have indeed been present within the genome for a long time, and even though copies that belong to a given family are similar in sequence, they are not identical because of evolutionary mechanisms that generate point mutations, rearrangements and indels. These mechanisms result in fragmented, divergent and mosaic copies that are difficult to identify by similarity approaches. Another biological characteristic that makes it difficult to identify their boundaries is that TEs sometimes insert preferentially into other TE copies to form nested elements (SanMiguel et al., 1996; Kaminker et al., 2002). Depending on the family, the number of copies of a TE can range from one or two copies for an ancient or not very active element to several million, as in the case of the SINEs in the human genome (The International Human Genome Sequencing Consortium, 2001). The number of occurrences of a given TE will depend on its activity in the genome, but also on the species analyzed. In the Drosophila genome, the most frequently occurring TE does not exceed hundreds of copies (Kaminker et al., 2002; Lerat et al., 2003) except the newly described DINE-1 elements that display thousand of non-autonomous copies (Kapitonov and Jurka, 2003). This means that the number of occurrence that can be expected in a genome is not constant. This impacts the parameters of a program, which will usually have to be adapted to suit the organisms being analyzed. One last problem that computational approaches have to deal with is the high cost in terms of calculation and memory size when analyzing very large genomes containing high occurrences of repeats.
Another problematic issue concerning TEs, once they have been detected, is to classify them into families and subfamilies. It is quite easy to identify the main classes of TEs, but as soon as we try to go further into a more detailed classification, automatic determination becomes a challenge. The first TEs were described on the basis of molecular biology analyses. Soon after, as a result of systematic searches in different organisms, their growing numbers allowed us to compare them with each other, and discover that different TEs can be phylogenetically related, which made further steps in classification possible. The methods of automatic detection have now made it possible to identify previously unknown elements. The new challenge we now face is how to situate these new elements relative to those already known. The accuracy of the links between TEs is particularly important if we are to understand their fate in genomes, and also to understand the dynamics of the genome itself.
I will try to review the methods currently available for the automatic annotation and classification of TEs in sequenced genomes as exhaustively as possible. I intend to highlight the main characteristics of the programs used, their main goals and the problems they can entail. I will also point out the various drawbacks of these different methods to help biologists who are unfamiliar with algorithmic methods to find their way through the dense forest of repeat identification methodology.
Programs intended to detect TEs and other repeats
The search for TEs and other repeats can be approached in several different ways. This depends on the level of knowledge of the repeats that is taken into account when identifying them in a genome sequence. It is possible to search for a specific element, to search for elements having particular structural features or to search for completely new and unknown elements solely on the basis of their repetitive nature. Table 1 shows the programs that have been developed to date according to the method they use. In two recent reviews, Bergman and Quesneville (2007) and Saha et al. (2008a) have described in greater detail the technical and algorithmic aspects of most of the programs mentioned here. I will not therefore insist on this aspect, but concentrate on describing how the programs are used in practice.
The library-based approaches: search by similarity of sequences
In these methods, the repetitive sequences are searched by comparing input data (a genome for example) to a set of reference sequences contained in a library. The library can either be homemade by the user, and tailored to the requirements and the question being asked, or it can be a generalist library, such as the commonly used REPBASE for instance (Jurka et al., 2005). This library contains curated consensus sequences of repeats from various eukaryotic organisms. The most extensively used library-based program is REPEATMASKER (Smit et al., 1996–2004). The program was originally designed to mask repeats in sequences to facilitate further investigation of aspects such as assembly and gene detection. It has become a gold standard for any search for repeats and TEs in genomes. The program performs a similarity search based on local alignments using one of the search engines CROSSMATCH or AB-BLAST. The output provided by the program proposes a detailed annotation of the repeats that have been detected, but also a modified version of the input sequences in which the repeats have been replaced by Ns. It has been extensively used alone in various different genome sequencing projects to identify repeats (Arabidopsis thaliana; The Arabidopsis Genome Initiative, 2000), the human genome (The International Human Genome Sequencing Consortium, 2001), the fugu fish (Aparicio et al., 2002), the mouse (The Mouse Genome Sequencing Consortium, 2002), the rice ssp indica (Yu et al., 2002) and ssp japonica (Goff et al., 2002) and the rat (The Rat Genome Sequencing Project Consortium, 2004). It has also been used in combination with other tools for use in the chicken (The International Chicken Genome Sequencing Consortium, 2004), the 12 Drosophila genomes (The Drosophila 12 Genomes Consortium, 2007) and Bos taurus (The Bovine Genome Sequencing and Analysis Consortium et al., 2009). Other tools apply the same kind of approach used in REPEATMASKER, such as CENSOR (Jurka et al., 1996), MASKERAID (Bedell et al., 2000), which is designed to enhance the performance of REPEATMASKER, PLOTREP (Tóth et al., 2006), and GREEDIER (Li et al., 2008). The PLOTREP program tries to deal with a recurrent problem that sometimes arises in similarity searches against a library, the fragmentation of some hits in the output. This fragmentation usually results from the presence of indels between the query and the match sequences, but can also be attributable to sequence divergence. After the similarity search step, PLOTREP finds matches that can be merged to form one single copy. This program has not yet been tested on genomic sequences, but the authors concluded that their tool should be able to identify full-length elements, even if they have been fragmented and disrupted. The GREEDIER program, in addition to finding fragmented repeats, also tries to detect nested elements. Li et al. (2008) have tested it on the Arabidopsis genome and the rice chromosome 10, to compare its performance with that of other tools using the same approach; they concluded that their program was an improvement over standard masking algorithms.
The REPEATMASKER program has been shown to be very efficient and fast. Moreover, it is particularly easy to use. The main drawback of programs based on a similarity search lies in the approach itself. As it is entirely based on homology, it obviously implies that this kind of method can only detect sequences that are already known to exist, and cannot detect completely novel elements. However, REPEATMASKER is often used as the first step in the identification of repeats, and also can be used in concordance with ab initio methods able to generate libraries of new repeats (see below). Moreover, this program is quite effective for finding low copy number families, which sometimes constitute an obstacle for ab initio methods.
The signature-based approaches: the search for particular features that characterize a given class of TEs
With this kind of approach, the program searches a query sequence for the occurrence of particular structures and motifs that are characteristic of a given type of repeat. This approach can be used to find new elements, but not new class of elements. The limitation of such approaches depends entirely on how much we know about the structure of elements belonging to particular classes, and also on the existence of characteristic structures. Some subclasses of elements are more highly structured than others, and this results in a bias toward detecting subclasses with evident structural characteristics rather than those with few or no conserved structures.
Programs for detecting non-LTR retrotransposons
The programs TSDFINDER (Szak et al., 2002), SINEDR (Tu et al., 2004) and RTANALYZER (Lucier et al., 2007) have been designed to detect non-LTR retrotransposons. The TSDFINDER program refines the coordinates of L1 insertions that are detected by REPEATMASKER. It first tests to see whether close matches can be merged, and it then searches for the presence of a polyA tail in 3′ of the sequence, and of target site duplications (TSDs) at both extremities of the copies, and finally, it detects any insertion and transduction events. A TSD consists of a short nucleotide sequence in the chromosome that is duplicated when the element is inserted. The authors of the program used it to analyze the recent L1 insertions in the human genome. The SINEDR program has been designed to detect known SINEs that are flanked by TSDs. The program has been shown to be able to identify a unique family of SINEs in the Aedes aegypti genome. The last program, RTANALYZER, has been designed to detect sequences of retrotransposed origin. It thus detects the signatures of L1 retrotranposition to find out whether the sequence analyzed has been retrotransposed by an L1. The signatures consist of the presence of TSDs, a polyA tail, and an endonuclease cleavage site in the 5′ end of the sequence. The program calculates a global retrotransposition score on the basis of the signatures detected. It has been implemented as a web application, and is intended for people working on mammalian genomes or gene sequences. Lucier et al. tested the program by using it to find retropseudogenes they had previously identified from human Y RNAs. The program slightly underestimated the number of retrotransposed hits.
Programs for detecting LTR retrotransposons
Several programs have been proposed for detecting new LTR retrotransposons in genomes. These programs, LTR_STRUC (McCarthy and McDonald, 2003), LTR_PAR (Kalyanaraman and Aluru, 2006), FIND_LTR (Rho et al., 2007), RETROTECTOR (Sperber et al., 2007), LTR_FINDER (Xu and Wang, 2007) and LTRHARVEST (Ellinghaus et al., 2008) are all based on a similar methodology. These programs take into account several structural features of LTR retrotransposons, such as a size range of the LTR sequences, the distance between the two LTRs of an element, the presence of TSDs at each extremity, the presence of critical sites for replication (the primer binding site and the polypurine tract) and the percentage identity between the two LTRs. Moreover, they can also rely on the presence of certain conserved motifs corresponding to the genes they encode. Some of the described features correspond to parameters that can be changed by the users in some programs.
To compare their ability to identify LTR retrotransposons, I tested all the programs on the X chromosome of D. melanogaster (http://www.hgdownload.cse.ucsc.edu/goldenPath/dm3/bigZips/). I decided to test these programs as their number was sufficient enough, their methodologies very close and they were all available with no major difficulty to run. Table 2 summarizes the results obtained for each program. The D. melanogaster genome is one of the most intensively annotated, and we possess the full annotation of its TEs. I did not test the RETROSPECTOR program, as it has been specifically designed to detect retroviral sequences in the human genome, whereas the other programs are more generalist in nature. On the X chromosome, 225 copies of LTR retrotransposons have been annotated. Among them, 96 correspond to full-length elements, the only kind of copies that the programs under investigation are able to detect according to their methodology. For each program, I computed the sensitivity, that is the percentage of LTR retrotransposons correctly identified. This corresponds to TP/(TP+FN), where TP is the number of true positives, which are the known repeats correctly identified by the tool, and FN is the number of false negatives, which are the known repeats not identified by the tool. It is not possible to compute the specificity, which is the proportion of true negatives identified. True negatives correspond to sequences known not to be LTR retrotransposons that are not identified as LTR retrotransposons by the tool. This proportion cannot be estimated in an ab initio approach.
The first program, LTR_STRUC, does not allow any change in its parameters. This program provided 70 candidates, 67 of which corresponded to annotated LTR retrotransposons. Two of the other three hits corresponded to LTR retrotransposons copies missed by the annotation, and also found by the other programs. Overall, this program gives quite good results, as it did not detect many false positives, however, it missed more than 30% of the elements.
The LTR_PAR program gives several results according to different level of confidence, 0 being the lowest level and 1 being the highest level. The level of confidence that gave the best results was level 0.5, which identified 41 copies. However, in each case, the number of false positive was very high, except for confidence level 1, but only 26 LTR retrotransposons were detected. The FIND_LTR program, using the default parameters, yielded 101 candidates, 84 of which corresponded to annotated LTR retrotransposons, and three to new LTR retrotransposons. In their article presenting the LTRHARVEST program, Ellinghaus et al. compared it with various other programs and tested different parameters. I run again the FIND_LTR program using the parameters proposed by Ellinghaus et al., and did indeed obtain fewer false positives. I did the same thing using the web application LTR_FINDER (default parameters and parameters proposed by Ellinghaus et al.). With the default parameters, the number of false positives was higher, but so was the number of true positives. The parameters proposed by Ellinghaus et al. led to the loss of nine true positives (72–63). The last program reviewed here, LTRHARVEST, gave very good results with the default parameters, with 94 true LTR retrotransposons detected. However, the number of false positives was particularly high (123 out of a total of 220 candidates). I also detected the three new LTR retrotransposons copies detected by FIND_LTR and by LTR_STRUC. Using the parameters proposed by the authors of the program to be used in Drosophila, the number of false positives decreased drastically (from 123 to 42), but remained high. Overall, LTRHARVEST and FIND_LTR gave the best results with regard to the number of true LTR retrotransposons detected. However, in each case, the number of false positives was very high, and performance depended considerably on the parameters selected for use.
The parameters that can be changed in the programs are usually the minimum and maximum length of the LTRs, the minimum and maximum distance between them and the minimum percentage of identity between them. These parameters are highly dependent on the organism in which the search is made. This implies that adjustments will always be needed when attempting to apply these programs on new organisms. It also implies that each candidate will need to be analyzed in detail, to make sure it is a true positive, when dealing with organisms for which there is no existing information about the LTR retrotransposon content.
Programs intended to detect MITEs
MITEs are a particular group of TEs that occur in genomes in high copy numbers (Wessler et al., 1995). They are short (<500 bp), possess terminal inverted repeats (TIRs) at their extremities, and transpose through a DNA intermediate. These elements are devoid of coding parts, and they depend on autonomous DNA transposons to be mobilized (Yang et al., 2009). Several programs have been designed to recognize these elements in genomes: FINDMITE (Tu, 2001), TRANSPO (Santiago et al., 2002), MITE Analysis Kit (MAK) (Yang and Hall, 2003) and MITE Uncovering SysTem (MUST) (Chen et al., 2009). The first program, FINDMITE, searches for potential MITEs that satisfy several criteria: particular TSDs, a certain length of TIRs and a minimum and maximum distance between TIRs. This program is able to find new elements, but cannot detect highly divergent copies. It was used on the Anopheles gambiae newly released genome, and detected eight new families of MITEs. The TRANSPO program is based on the detection of TIRs from a query sequence. This means that it cannot find new elements, but unlike FINDMITE, it can detect old copies. It was used to perform a genome-wide analysis of a particular MITE family in the A. thaliana genome. The MAK tool kit groups programs used to automate MITE analysis. From a given MITE sequence, it can retrieve the sequences of other members of the family, identify the neighboring genes and can predict the anchor elements, that is the autonomous elements responsible for the transposition of the MITE. This program is therefore able to find new members of a known family, but can also detect new members of related families. Tested on the Arabidopsis genome, it identified two new families. The MUST program takes an approach that is also based on the detection of TIRs. It searches a genome for all the occurrence of TIRs in a window of a given size (500 bp by default), and for TSDs around them. It then uses a method based on sequence alignment to confirm or reject, and to classify candidate MITEs. Chen et al. tested their program on two bacterial genomes, in which it identified hundreds of candidates. The authors temper their finding by pointing out the necessity for manual verification to eliminate potential false positives. I intended to test these programs but the provided URL of one of the five programs (MAK) does not seem to be valid and prevented me to download it, and the FINDMITE program gave an error when I tried to run it.
A program for detecting helitrons
One program has been recently proposed to detect helitrons in genomes (Du et al., 2008). Helitrons are a new class of TEs found in animals and plants (Kapitonov and Jurka, 2001). These elements have basic features such as conserved short sequences in their 5′ and 3′ extremities, palindromes of 16–20 bp corresponding to hairpin loops near the 3′ end, and flanking A and T host nucleotides at the 5′ and 3′ ends, respectively. The HELITRONFINDER program is dedicated exclusively to predicting the HelA type, a particular class of helitron found in maize, by searching in the maize genome sequence for its pattern using the regular expression abilities of the program language used.
The de novo approaches: search for repeats of any kind
The idea of de novo approaches is to take advantage of the repetitive nature of TEs and other repeats, without relying on repetitive elements or motifs that are already known. These approaches are intended mainly to discover new repeats, and are becoming particularly valuable as the number of sequenced genomes increases about which we have little or no information concerning their repeat content. These approaches use different kinds of methodology, and their final goals may also differ. Some methods are designed to provide an exhaustive list of repeats in the genome, whereas others (sometimes in addition to doing this) are intended to define families of repeats, sometimes constructing consensus sequences for each family that can subsequently be used as a reference, using REPEATMASKER, for example, to search for the positions and occurrences of these repeats in the genome. There are two main approaches to detecting repeats in a sequence. The first consists of comparing a sequence with itself, and the second consists of searching for the repeated occurrence of small words (known as k-mers), and this can be extended to larger sequences.
Self-comparison approaches
The self-comparison approaches are used by the REPEAT PATTERN TOOLKIT (Agarwal and States, 1994), RECON (Bao and Eddy, 2002), PILER (Edgar and Myers, 2005) and the BLASTER suite (used in Quesneville et al., 2005).
The REPEAT PATTERN TOOLKIT was the first attempt to detect repeats using this method. The approach is based on a sequence similarity scoring system, and uses BLAST (Altschul et al., 1990) to perform the self-comparison. The grouping of repeats is then formed by clustering. The program was originally tested on chromosome III of Caenorhabditis elegans, which was the longest available contiguous segment of DNA at the time. Agarwal and States showed that it contains 12% of repeats, which is congruent with the estimated amount for the entire genome (Stein et al., 2003).
The RECON program is one of the most used programs, and it also uses the BLAST program to perform the self-comparison, followed by a clustering method to form repeat families. The method has been tested on a random 3 Mb sample of the human genome (corresponding to 0.1% of the complete genome). It was more recently used to identify repeats in a nematode genome, Ancylostoma caninum (Abubucker et al., 2008), and in the chicken genome, where it was used in addition to REPEATMASKER (The International Chicken Genome Sequencing Consortium, 2004).
The PILER program uses another tool to perform the self-alignments called PALS (Pairwise Alignment of Long Sequences). This program identifies certain alignments that form characteristic patterns of a given repeat type to increase the reliability. It distinguishes between the tandem arrays (PILER-TA), which correspond to the satellites, the dispersed families (PILER-DF), which correspond to the TEs, the pseudosatellites (PILER-PS) and the terminal repeats (PILER-TR). A consensus sequence is then generated after multiple alignments of all the members of a family, and this consensus can then be used in a REPEATMASKER search, for example. The program has been tested to identify satellites and pseudosatellites in the Arabidopsis and human genomes, and to identify gypsy like elements in D. melanogaster. It has recently been used to search for repeats in B. taurus (The Bovine Genome Sequencing and Analysis Consortium et al., 2009), and in the 12 Drosophila genomes (The Drosophila 12 Genomes Consortium, 2007), where it was used in addition to other programs, and in the bat genome Myotis lucifugus (Ray et al., 2007).
In the BLASTER suite, the BLAST program is also used, and then two other programs (MATCHER and GROUPER) are used to map the matches on the genome and cluster the sequences into families. The program has been used by its original authors in several studies in insects and plants, and particularly in D. melanogaster (Quesneville et al., 2005).
k-mer and spaced seed approaches
There are numerous programs based on k-mer approach or on its derivative, the spaced seed approach. In the k-mer method, a repeat is viewed like a substring of length k that occurs more than once in a sequence. The matches have to be identical. The space seed approach is an extension of the k-mer approach, and allows some variations in the sequence of the seed, such as the percentage identity and the length. The programs that use one or other of these methods are as follows: REPUTER (Kurtz and Schleiermacher, 1999), VMATCH (Kurtz, unpublished), REPEAT-MATCH (Delcher et al., 1999), MER-ENGINE (Healy et al., 2003), FORREPEATS (Lefebvre et al., 2003), REAS (Li et al., 2005), REPEATSCOUT (Price et al., 2005), RAP (Campagna et al., 2005), REPSEEK (Achaz et al., 2007), TALLYMER (Kurtz et al., 2008) and P-CLOUDS (Gu et al., 2008).
REPUTER was one of the first programs to apply the k-mer approach. The algorithm is based on a suffix tree data structure. This structure contains all suffixes than can be degenerated from any string. It makes it possible to determine all the exact repetitive substrings in a complete genome. Although the program was not tested on genomic data by the original authors, it has been used in various studies to detect TEs (in Ophiostoma ulmi and O. novo-ulmi, agents of the Dutch elm disease (Bouvet et al., 2006), Medicago truncatula and Lotus japonicus (Cannon et al., 2006)). This kind of approach is also found in REPEAT-MATCH and in VMATCH, which is the program that subsumes REPUTER. The MER-ENGINE tool was designed to annotate any sequences rapidly by counting its constituent words, and was not originally intended for use in searching specifically for repeats. The original authors tested their program on the human genome but found significant discordance between annotated repeats and their regions because of the fact that the program cannot find diverged repeats. However, they conclude that the program would be sufficient enough to design probes. The FORREPEATS program is based on a data structure known as factor oracle. It first detects exact repeats in a sequence, and then computes approximate repeats and performs pairwise comparison. The original authors, Lefebvre et al., tested their program on the genome of A. thaliana. The REAS, REPEATSCOUT and TALLYMER programs all build a library of high-frequency, fixed length, k-mers and use them as seeds to define the family of repeats. A particular feature of REAS is that the program is designed to work on sequencing reads rather than on assembled sequences. It was tested on the japonica rice genome. The results gave more than 8000 TE candidates, more than 1200 of which matched known TEs in REPBASE, 707 of the candidates matched TE-related proteins, but the remainder could not be classified and were mainly false positives. The REAS program has also been used with other programs to detect TEs in the 12 Drosophila genomes (The Drosophila 12 genomes consortium, 2007), and in the new assembly of the Bombyx mori genome (The International Silkworm Genome Consortium, 2008). The TALLYMER program was tested on maize BAC sequences, and the results compared to masking by REPEATMASKER. The two methods gave similar results. The RAP and REPSEEK programs detect approximate repeats in the genome rather than exact repeats. Both programs were evaluated on the C. elegans genome. RAP found some new regions of repeats that correspond to duplicated genes, whereas REPSEEK results showed that 15% of the repeats were not found by REPEATMASKER. The approach of P-CLOUD is based on the hypothesis that repeated elements are grouped into clusters of similar oligos, and that it should be statistically possible to detect clusters of relative oligos. Using this approach, Gu et al., evaluated the repeat content of chromosomes 1 and X of Homo sapiens. The results showed that 50.7% of the sequence was recognized by the program as repeats. Among them, 14.7% were not found by REPEATMASKER, indicating that these sequences may not in fact be TEs, but members of multigenic families, pseudogenes or the result of segmental duplication.
Identification of repeat families
Some of the programs I have already mentioned involve the clustering of repeats into families, whereas others are mainly designed to built repeat families. Of these latter programs, some use tools that detect repeats and then propose other way of defining the repeat families. This is the case for REPEATFINDER (Volfovsky et al., 2001), which uses either REPUTER or REPEAT-MATCH to define exact repeats as the basis for constructing classes of repeats. It then merges different exact repeats that are close or that overlap. The program was evaluated by its original authors in various organisms: several bacterial genomes and the Arabidopsis and rice genomes. The program showed that most of the detected repeats result from duplication rather than TEs. The REPEATGLUER program (Pevzner et al., 2004) is based on a de Bruijn graph to represent the repeats. This graph represents every k-mer in a genome sequence as a node. It then connects two nodes by a directed edge if they are overlapping in the genome. A consensus sequence is built inside each family constructed, and the number of occurrences is determined. The program has been developed to enhance the EULER assembler (Pevzner et al., 2001).
Evaluating the prediction programs
The large number of methods available for the de novo prediction of repeats makes it necessary to evaluate these approaches, as they cannot be compared solely on the basis of their published description. Indeed, the evaluation of programs by their original authors is usually carried out on different organisms, and the way the results or the data used to compare them are presented, makes it difficult to appreciate the objective capacities of the different programs. An empirical test of some of these tools has been performed by Saha et al. (2008b). They selected six of the most popular and widely used programs: RECON, REAS, REPEATGLUER, REPEATSCOUT, REPEATFINDER and PILER. Each program was tested on the same data set: rice chromosome 12, which is the chromosome with the highest repeat content in this genome. They evaluated each program on the basis of its computing time, its effectiveness for finding known repeats, its capacity to find new repeats and its ability to identify different types of repeats. They estimated that REAS was the best program for use on unassembled sequence reads, even though it found fewer novel repeats than RECON, and that REPEATSCOUT gave the best results for assembled genomic sequences. They pointed out that some programs produced incoherent results, such as REPEATGLUER, which seemed to show that the data set consisted almost entirely of repeats! The PILER program missed a lot a known repeats, but was one of the fastest programs. REPEATFINDER found a lot of novel repeats, but some of them may have been false positives. Overall, Saha et al. showed that there is considerably variation in the performance of the programs they tested, and that further improvements are essential. They also pointed out some of the problems that I will discuss in the last part of this review.
Other kind of approaches
Some studies have used other kind of approaches to detect repeats. One very interesting approach has been proposed by Caspi and Pachter (2006). In their method, the authors proposed that by aligning the genomes of closely related species, it would be possible to identify TE insertions that are present in one genome but not in the other(s), as it would result in a large gap in the alignment in the other species sequences. This method makes it possible to detect new insertions, and also to date the corresponding insertion events. One drawback of this approach is that it is very dependent on the quality of the genome alignments, but it also requires the use of sufficiently closely related species. Other methods that have been proposed have attempted to take into account some global particularities of TEs, such as the nucleotide composition, arguing that the base composition of TEs differs from that of the host genes. Andrieu et al. (2004) have developed a method based on a Hidden Markov Model that can be applied on whole genome. This method requires good training data sets, and is also very dependent on the base composition of the genome. It has been shown that TEs, of whatever species, are AT-rich (Lerat et al., 2000, 2002), which implies that this method would work better in genomes that are GC-rich rather than in AT-rich genomes. The method also implies having to determine the training set all over again for each new genome analyzed. Another method, which is completely different from the previous one, is based on a Fourier transform. The spectral repeat finder (Sharma et al., 2004) analyzes a sequence to identify the length of potential repeats by evaluating the power spectrum, which is a Fourier transformation of a sequence of variables in the ‘frequency domain’. Each periodic signal (repeats in a sequence) is evidenced as a peak in the power spectrum. High intensity peaks in the power spectrum represent candidates that can be used as seeds to perform local alignment search to detect similar elements and construct a consensus sequence. The greater the number of repeats, the stronger the peaks, which means that this method should work very well for detecting exact tandem repeats.
Some other programs have been developed that are dedicated to the detection of repeats other than TEs. Tandem Repeats Finder (TRF) (Benson, 1999), Tandem Repeat Occurrence Locator (TROLL) (Castelo et al., 2002), MREPS (Kolpakov et al., 2003), TRAP (Sobreira et al., 2006) and Optimized Moving Window Spectral Analysis (OMWSA) (Du et al., 2007) have been developed specifically to detect tandem repeats. The Inverted Repeat Finder (IRF) program (Warburton et al., 2004) was designed to search for inverted repeats.
Classification of repeats into families
The goal of some programs is the automation of the classification of repeats once they have been identified, as it is a very long and difficult process. LTR-MINER (Pereira, 2004) uses the output of REPEATMASKER to identify both complete LTR retrotransposons and solo-LTRs. The RETROMAP program iteratively searches for reverse transcriptions to define LTR retrotransposon insertions using the output of a BLAST search (Peterson-Burch et al., 2004). The DOMAINORGANIZER tool has been designed to classify elements based on the combinations of elementary domains that are characteristic of a given family (Tempel et al., 2006). These domains are defined as conserved segments in multiple alignments. The TECLASS program is more generalist, and intends to classify repeats according to the main classes of elements (Abrusán et al., 2009). It is based on an approach of machine learning that uses the oligomer frequencies of the repeats. In REPCLASS, the tool uses different approaches to automatically annotate TEs through three modules (Feschotte et al., 2009). One module involves a homology approach, the second a structural approach that searches for structural features characteristic of different classes of elements and the third is based on a search for TSDs. The results of each of the modules are then combined. All these programs constitute the last step before analyzing the repeat content of a genome.
Grouping different programs: a pipeline of programs
Given the number of different programs, all of which have their own qualities and drawbacks, pipelines have been developed that include several of the programs I have already mentioned. Generally a pipeline is developed to answer a particular question. The REPEATMODELER pipeline (Smit, unpublished http://www.repeatmasker.org/RepeatModeler.html) includes the programs RECON, REPEATSCOUT, REPEATMASKER and TRF. It uses the output of the RECON and REPEATSCOUT programs to build, refine and classify consensus models of putative interspersed repeats. Quesneville et al. (2005) proposed a ‘combined evidence’ approach to try to increase the quality of TE annotations at the same level as the gene annotation. To do this, they designed the REPET pipeline, which integrates the findings of homology-based and de novo repeat identification methods (BLASTER, RECON, REPEATMASKER, TRF and MREPS). They tested their pipeline on the D. melanogaster genome, which at the time was the best annotated. Their work added to the number of annotated TEs in this genome. With the aim of improving the annotation of TEs in Dipterans, Smith et al. (2007) used a pipeline known as REPEATRUNNER, that uses PILER, REPEATMASKER and BLASTX. Their analysis of the D. melanogaster genome also increased the proportion of annotated TEs, and provided information about the TE content of the other sequenced Drosophila genomes and of the A. gambiae genomes. To perform an evolutionary analysis of the TEs on mammalian genomes, Giordano et al. (2007), have developed a package called TRANSPOSON CLUSTER FINDER that can be used to defragment TEs and to identify TEs inserted into each other. To do this, the program uses the output of REPEATMASKER. This tool can be used to establish the chronological order of TE insertions into the human genome. The REANNOTATE tool also uses the output of REPEATMASKER (Pereira, 2008). It also used the same approach of defragmenting elements, resolving the chronological order of the insertion and estimating the age of the LTR retrotransposons. The program has been applied to the human genome. The TENEST program was also developed to determine the chronological order of TE insertions, and to make it possible to visualize nested elements in plants (Kronmiller and Wise, 2008). It uses the output of BLAST after a comparison of a repeat database and the genome. The DAWGPAWS pipeline (Estill and Bennetzen, 2009) is dedicated to the annotation of genes and TEs in plant genomes. It uses several programs (LTR_STRUC, LTR_FINDER, LTR_PAR, FIND_LTR, FINDMITE, TRF, REPSEEK, REPEATMASKER and TENEST). The RETROPRED tool was designed by integrating PALS, PILER, MEME and ANN to find particular non-LTR retrotransposons (Naik et al., 2008). By a sequence homology approach, the TARGET (Tree Analysis of Related Genes and Transposons) pipeline was designed using Blast, the multiple sequence alignment tool MUSCLE (Edgar, 2004) and tree reconstruction to characterize not only TEs but also gene families (Han et al., 2009). This tool is available as a web interface. The REREP (Read Repeat Finder) pipeline has been designed to help to identify repetitive units before the assembly phase of a genome (Otto et al., 2008). The program was tested on one cosmid of Leishmania major, and the sequences from the genome survey sequencing of L. braziliensis, which corresponds to 1.4% of the complete genome.
The problems underlying the programs for identifying TEs and repeats
The existence of programs able to detect repeats and TEs in genomes raises particular problems, some of which have already been pointed out by Saha et al. (2008b). The first difficulty is to be able to appreciate the value of these numerous programs, as they have not always been cross-tested by different researchers. However, some more specific problems arise from very trivial issues. I tried to determine how easy the different programs are to use by retrieving them, installing them and running them. Some programs are not provided as a downloadable archive, but require a direct request to the authors, who do not always respond. For instance, it was not possible to get hold of the RAP program. Even with Web site address, some programs were not possible to be downloaded because the provided address was no longer valid, like for the MAK or the LTR_MINER programs. Furthermore, some of these programs were the product of a short-term research project, and are no longer maintained. As some of these programs rely on other tools that may have evolved independently, especially their output format, so the program will need to adapt. This problem arose with the REPEATFINDER program, which relies on REPUTER output. The output file format of the last program has changed since REPEATFINDER was published, making this program unusable without tinkering with the code. Saha et al. (2008b) had already brought this issue to light, reporting that they had had to modify some of the programs they tested to make them work. This indicates that the average biologist would not be able to run most of these programs. This issue is also related to the fact that very few programs offer any detailed documentation to help the user to install the program, make it run, let alone modify it. Even with programs that can be downloaded, sometimes parts of the program seem to be missing, and the user has to contact the authors to get hold of them, as it was the case for the FIND_LTR program. A problem also occurred when I tried to use REAS, for which the compilation was particularly fastidious, and necessitated making numerous corrections to the codes. In the end, the program still failed to run, because of a segmentation fault that would have required a detailed inspection of the codes of the program, and for which the authors were of no help. These problems obviously compromise the use of the tools concerned. They reflect either the wish of their authors to control the use of the programs, or in some cases just that these programs correspond to work done at a given time, with no long-term perspective in view. Most of these programs were intended to answer a question that arose at a specific time. The trouble is that rather than trying to use tools that already exist, some authors prefer to develop their own. Developing new softwares when some already exists that could do the job would be justifiable if the aim was to improve the method. But this is rarely the case. However, this determination to make new tools probably also arises precisely from the fact that the existing programs are not easy to use!
Another problem arises when trying to use the programs for data of a different kind from that tested in the original publication. The parameters are not always appropriate for all kinds of data. This was revealed by the empirical test carried out by Saha et al. (2008b), and also by the comparison of LTRHARVEST with similar programs by its authors (Ellinghaus et al., 2008). When I tested tools intended for finding LTR retrotransposons this problem occurred with LTR_PAR, which did not produce any coherent result for the X chromosome of D. melanogaster, whereas it did work for the yeast genome, the organism on which the program was originally developed. The only way to get results was to contact the author, who was able to produce coherent results, although without providing any explanation for my failure. The need to find the right parameters is a real challenge, especially if the programs are intended for use in detecting de novo TEs. When a new organism is sequenced, with repeats of which we know nothing, it is particularly difficult to decide which would be the best parameters to use to detect them. Moreover, some programs published have not been tested on real data. When this is the case, the results are rarely compared to well-curated annotations that would help to validate the functionality of the program.
With the ever-increasing number of programs comes the need to test them objectively to identify the ones that look most interesting to maintain and develop. There is also a great need to provide users with better information and documentation. Biologists are the only people who can confirm whether the findings of these programs are trustworthy, but very few have been made accessible to people with the level of informatics skills usually possessed by biologists. Detecting repeats in genomes is indeed a major challenge in informatics, but the biological question behind this has to remain the main objective.
Conclusion
The question of which program to use arises from the large number of programs that claim to detect repeats and TEs in genomes. Saha et al. (2008a) and Bergman and Quesneville (2007) suggest that no single program could be sufficiently exhaustive to detect all repeats. This implies that using several different programs and carrying out a cross comparison of their results has the best chance of finding reliable results as any single program. However, this makes it indispensable to test the results provided by each program independently, and not simply rely on the claims made by their authors. The ideal solution would be to test all programs against the same data set to obtain a true comparison of how they perform, but this would demand a huge amount of work and the task is not facilitated by the difficulties encountered in using the programs that are already available, and by the fact that new programs are constantly being published.
References
Abrusán G, Grundmann N, DeMester L, Makalowski W (2009). TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25: 1329–1330.
Abubucker S, Martin J, Yin Y, Fulton L, Yang SP, Hallsworth-Pepin K et al. (2008). The canine hookworm genome: analysis and classification of Ancylostoma caninum survey sequences. Mol Biochem Parasitol 157: 187–192.
Achaz G, Boyer F, Rocha EP, Viari A, Coissac E (2007). Repseek, a tool to retrieve approximate repeats from large DNA sequences. Bioinformatics 23: 119–121.
Agarwal P, States DJ (1994). The Repeat Pattern Toolkit (RPT): analyzing the structure and evolution of the C. elegans genome. Proc Int Conf Intell Syst Mol Biol 2: 1–9.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990). Basic local alignment search tool. J Mol Biol 215: 403–410.
Andrieu O, Fiston AS, Anxolabéhère D, Quesneville H (2004). Detection of transposable elements by their compositional bias. BMC Bioinformatics 5: 94.
Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P et al. (2002). Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science 297: 1301–1310.
Bao Z, Eddy SR (2002). Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res 12: 1269–1276.
Bedell JA, Korf I, Gish W (2000). MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 16: 1040–1041.
Benson G (1999). Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27: 573–580.
Bergman CM, Quesneville H (2007). Discovering and detecting transposable elements in genome sequences. Brief Bioinform 8: 382–392.
Biémont C, Vieira C (2006). Junk DNA as an evolutionary force. Nature 443: 521–524.
Bouvet GF, Jacobia V, Bernier L (2006). Characterization of three DNA transposons in the Dutch elm disease fungi and evidence of repeat-induced point (RIP) mutations. Fungal Genet Biol 44: 430–443.
Britten RJ, Kohne DE (1968). Repeated sequences in DNA. Hundreds of thousands of copies of DNA sequences have been incorporated into the genomes of higher organisms. Science 161: 529–540.
Campagna D, Romualdi C, Vitulo N, Del Favero M, Lexa M, Cannata N et al. (2005). RAP: a new computer program for de novo identification of repeated sequences in whole genomes. Bioinformatics 21: 582–588.
Cannon SB, Sterck L, Rombauts S, Sato S, Cheung F, Gouzy J et al. (2006). Legume genome evolution viewed through the Medicago truncatula and Lotus japonicus genomes. Proc Natl Acad Sci USA 103: 14959–14964.
Capy P, Bazin C, Higuet D, Langin T (1997). Dynamics and Evolution of Transposable Elements. R.G. Landes company: Austin, TX.
Caspi A, Pachter L (2006). Identification of transposable elements using multiple alignments of related genomes. Genome Res 16: 260–270.
Castelo AT, Martins W, Gao GR (2002). TROLL—tandem repeat occurrence locator. Bioinformatics 18: 634–636.
Chen Y, Zhou F, Li G, Xu Y (2009). MUST: a system for identification of miniature inverted-repeat transposable elements and applications to Anabaena variabilis and Haloquadratum walsbyi. Gene 436: 1–7.
Delcher AL, Kasif S, Fleischmann RD, Peterson J, White O, Salzberg SL (1999). Alignment of whole genomes. Nucleic Acids Res 27: 2369–2376.
Du C, Caronna J, He L, Dooner HK (2008). Computational prediction and molecular confirmation of Helitron transposons in the maize genome. BMC Genomics 9: 51.
Du L, Zhou H, Yan H (2007). OMWSA: detection of DNA repeats using moving window spectral analysis. Bioinformatics 23: 631–633.
Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32: 1792–1797.
Edgar RC, Myers EW (2005). PILER: identification and classification of genomic repeats. Bioinformatics 21: i152–i158.
Ellinghaus D, Kurtz S, Willhoeft U (2008). LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatic 9: 18.
Estill JC, Bennetzen JL (2009). The DAWGPAWS pipeline for the annotation of genes and transposable elements in plant genomes. Plant Methods 5: 8.
Feschotte C, Keswani U, Ranganathan N, Guibotsy ML, Levine D (2009). Exploring repetitive DNA landscape using REPCLASS, a tool that automates the classification of transposable elements in eukaryotic genomes. Genome Biol Evol 2009: 205–220.
Finnegan DJ (1989). Eukaryotic transposable elements and genome evolution. Trends Genet 5: 103–107.
Giordano J, Ge Y, Gelfand Y, Abrusán G, Benson G, Warburton PE (2007). Evolutionary history of mammalian transposons determined by genome-wide defragmentation. PLoS Comput Biol 3: e137.
Goff SA, Ricke D, Lan TH, Presting G, Wang R, Dunn M et al. (2002). A draft sequence of the rice genome (Oryza sativa L. ssp. Japonica). Science 296: 92–100.
Gu W, Castoe TA, Hedges DJ, Batzer MA, Pollock DD (2008). Identification of repeat structure in large genomes using repeat probability clouds. Anal Biochem 380: 77–83.
Han Y, Burnette III JM, Wessler SR (2009). TARGeT: a web-based pipeline for retrieving and characterizing gene and transposable element families from genomic sequences. Nucleic Acids Res 37: e78.
Healy J, Thomas EE, Schwartz JT, Wigler M (2003). Annotating large genomes with exact word matches. Genome Res 13: 2306–2315.
Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J (2005). Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res 110: 462–467.
Jurka J, Klonowski P, Dagman V, Pelton P (1996). CENSOR—a program for identification and elimination of repetitive elements from DNA sequences. Comput Chem 20: 119–121.
Kalyanaraman A, Aluru S (2006). Efficient algorithms and software for detection of full-length LTR retrotransposons. J Bioinform Comput Biol 4: 197–216.
Kaminker JS, Bergman CM, Kronmiller B, Carlson J, Svirskas R, Patel S et al. (2002). The transposable elements of the Drosophila melanogaster euchromatin: a genomics perspective. Genome Biol 3: RESEARCH0084.
Kapitonov VV, Jurka J (2001). Rolling-circle transposons in eukaryotes. Proc Natl Acad Sci USA 98: 8714–8719.
Kapitonov VV, Jurka J (2003). Molecular paleontology of transposable elements in the Drosophila melanogaster genome. Proc Natl Acad Sci USA 100: 6569–6574.
Kidwell MG, Lisch DR (2001). Transposable elements, parasitic DNA, and genome evolution. Evolution 55: 1–24.
Kim JM, Vanguri S, Boeke JD, Gabriel A, Voytas DF (1998). Transposable elements and genome organization: a comprehensive survey of retrotransposons revealed by the complete Saccharomyces cerevisiae genome sequence. Genome Res 8: 464–478.
Kolpakov R, Bana G, Kucherov G (2003). mreps: efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res 31: 3672–3678.
Kronmiller BA, Wise RP (2008). TEnest: automated chronological annotation and visualization of nested plant transposable elements. Plant Physiol 146: 45–59.
Kurtz S, Narechania A, Stein JC, Ware D (2008). A new method to compute K-mer frequencies and its application to annotate large repetitive plant genomes. BMC Genomics 9: 517.
Kurtz S, Schleiermacher C (1999). REPuter: fast computation of maximal repeats in complete genomes. Bioinformatics 15: 426–427.
Lefebvre A, Lecroq T, Dauchel H, Alexandre J (2003). FORRepeats: detects repeats on entire chromosomes and between genomes. Bioinformatics 19: 319–326.
Lerat E, Biémont C, Capy P (2000). Codon usage and the origin of P elements. Mol Biol Evol 17: 467–468.
Lerat E, Capy P, Biémont C (2002). Codon usage by transposable elements and their host genes in five species. J Mol Evol 54: 625–637.
Lerat E, Rizzon C, Biémont C (2003). Sequence divergence within transposable element families in the Drosophila melanogaster genome. Genome Res 13: 1889–1896.
Li RQ, Ye J, Li S, Wang J, Han Y, Ye C et al. (2005). ReAS: recovery of ancestral sequences for transposable elements from the unassembled reads of a whole genome shotgun. PLoS Comput Biol 1: e43.
Li X, Kahveci T, Settles AM (2008). A novel genome-scale repeat finder geared towards transposons. Bioinformatics 24: 468–476.
Lucier JF, Perreault J, Noël JF, Boire G, Perreault JP (2007). RTAnalyzer: a web application for finding new retrotransposons and detecting L1 retrotransposition signatures. Nucleic Acids Res 35: W269–W274.
McCarthy EM, McDonald JF (2003). LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics 19: 362–367.
Naik PK, Mittal VK, Gupta S (2008). RetroPred: a tool for prediction, classification and extraction of non-LTR retrotransposons (LINEs & SINEs) from the genome by integrating PALS, PILER, MEME and ANN. Bioinformation 2: 263–270.
Otto TD, Gomes LH, Alves-Ferreira M, de Miranda AB, Degrave WM (2008). ReRep: computational detection of repetitive sequences in genome survey sequences (GSS). BMC Bioinformatics 9: 366.
Pereira V (2004). Insertion bias and purifying selection of retrotransposons in the Arabidopsis thaliana genome. Genome Biol 5: R79.
Pereira V (2008). Automated paleontology of repetitive DNA with REANNOTATE. BMC Genomics 9: 614.
Peterson-Burch BD, Nettleton D, Voytas DF (2004). Genomic neighborhoods for Arabidopsis retrotransposons: a role for targeted integration in the distribution of the Metaviridae. Genome Biol 5: R78.
Pevzner PA, Tang H, Tesler G (2004). De novo repeat classification and fragment assembly. Genome Res 14: 1786–1796.
Pevzner PA, Tang H, Waterman M (2001). An Eulerian path approach to DNA fragment assembly. Proc Nath Acad Sci USA 98: 9748–9753.
Price AL, Jones NC, Pevzner PA (2005). De novo identification of repeat families in large genomes. Bioinformatics 21: i351–i358.
Quesneville H, Bergman CM, Andrieu O, Autard D, Nouaud D, Ashburner M et al. (2005). Combined evidence annotation of transposable elements in genome sequences. PLoS Comput Biol 1: 166–175.
Ray DA, Pagan HJ, Thompson ML, Stevens RD (2007). Bats with hATs: evidence for recent DNA transposon activity in genus Myotis. Mol Biol Evol 24: 632–639.
Rho M, Choi J-H, Kim S, Lynch M, Tang H (2007). De novo identification of LTR retrotransposons in eukaryotic genomes. BMC Genomics 8: 90.
Saha S, Bridges S, Magbanua ZV, Peterson DG (2008a). Computational approaches and tools used in identification of dispersed repetitive DNA sequences. Trop Plant Biol 1: 85–96.
Saha S, Bridges S, Magbanua ZV, Peterson DG (2008b). Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res 36: 2284–2294.
SanMiguel P, Gaut BS, Tikhonov A, Nakajima Y, Bennetzen JL (1998). The paleontology of intergene retrotransposons of maize. Nat Genet 20: 43–45.
SanMiguel P, Tikhonov A, Jin YK, Motchoulskaia N, Zakharov D, Melake-Berhan A et al. (1996). Nested retrotransposons in the intergenic regions of the maize genome. Science 274: 765–768.
Santiago N, Herráiz C, Goñi JR, Messeguer X, Casacuberta JM (2002). Genome-wide analysis of the Emigrant family of MITEs of Arabidopsis thaliana. Mol Biol Evol 19: 2285–2293.
Sharma D, Issac B, Raghava GP, Ramaswamy R (2004). Spectral Repeat Finder (SRF): identification of repetitive sequences using Fourier transformation. Bioinformatics 20: 1405–1412.
Smit AFA, Hubley R, Green P (1996–2004). RepeatMasker Open-3.0. (http://www.repeatmasker.org).
Smith CD, Edgar RC, Yandell MD, Smith DR, Celniker SE, Myers EW et al. (2007). Improved repeat identification and masking in Dipterans. Gene 389: 1–9.
Sobreira TJ, Durham AM, Gruber A (2006). TRAP: automated classification, quantification and annotation of tandemly repeated sequences. Bioinformatics 22: 361–362.
Sperber GO, Airola T, Jern P, Blomberg J (2007). Automated recognition of retroviral sequences in genomic data—RetroTector. Nucleic Acids Res 35: 4964–4976.
Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N et al. (2003). The genome sequence ofs Caenorhabditis briggsae: a platform for comparative genomics. PLoS Biol 1: E45.
Szak ST, Pickeral OK, Makalowski W, Boguski MS, Landsman D, Boeke JD (2002). Molecular archeology of L1 insertions in the human genome. Genome Biol 3: research0052.
Tang H (2007). Genome assembly, rearrangement, and repeats. Chem Rev 107: 3391–3406.
Tempel S, Giraud M, Lavenier D, Lerman IC, Valin AS, Couée I et al. (2006). Domain organization within repeated DNA sequences: application to the study of a family of transposable elements. Bioinformatics 22: 1948–1954.
The Arabidopsis Genome Initiative (2000). Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815.
The Bovine Genome Sequencing and Analysis Consortium (2009). The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science 324: 522–528.
The Drosophila 12 Genomes Consortium (2007). Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218.
The International Chicken Genome Sequencing Consortium (2004). Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432: 695–716.
The International Human Genome Sequencing Consortium (2001). Initial sequencing and analysis of the human genome. Nature 409: 860–921.
The International Silkworm Genome Consortium (2008). The genome of a lepidopteran model insect, the silkworm Bombyx mori. Insect Biochem Mol Biol 38: 1036–1045.
The Mouse Genome Sequencing Consortium (2002). Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562.
The Rat Genome Sequencing Project Consortium (2004). Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 428: 493–521.
Tóth G, Deák G, Barta E, Kiss GB (2006). PLOTREP: a web tool for defragmentation and visual analysis of dispersed genomic repeats. Nucleic Acids Res 34: W708–W713.
Tu Z (2001). Eight novel families of miniature inverted repeat transposable elements in the African malaria mosquito, Anopheles gambiae. Proc Natl Acad Sci USA 98: 1699–1704.
Tu Z, Li S, Mao C (2004). The changing tails of a novel short interspersed element in Aedes aegypti: genomic evidence for slippage retrotransposition and the relationship between 3′ tandem repeats and the poly(dA) tail. Genetics 168: 2037–2047.
Volfovsky N, Haas BJ, Salzberg SL (2001). A clustering method for repeat analysis in DNA sequences. Genome Biol 2: RESEARCH0027.
Warburton PE, Giordano J, Cheung F, Gelfand Y, Benson G (2004). Inverted repeat structure of the human genome: the X-chromosome contains a preponderance of large, highly homologous inverted repeats that contain testes genes. Genome Res 14: 1861–1869.
Wessler SR, Bureau TE, White SE (1995). LTR-retrotransposons and MITEs: important players in the evolution of plant genomes. Curr Opin Genet Dev 5: 814–821.
Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B et al. (2007). A unified classification system for eukaryotic transposable elements. Nat Rev Genet 8: 973–982.
Xu Z, Wang H (2007). LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res 35: W265–W268.
Yang G, Hall TC (2003). MAK, a computational tool kit for automated MITE analysis. Nucleic Acids Res 31: 3659–3665.
Yang G, Nagel DH, Feschotte C, Hancock CN, Wessler SR (2009). Tuned for transposition: molecular determinants underlying the hyperactivity of a Stowaway MITE. Science 325: 1391–1394.
Yu J, Hu S, Wang J, Wong GK, Li S, Liu B et al. (2002). A draft sequence of the rice genome (Oryza sativa L. ssp. Indica). Science 296: 79–92.
Acknowledgements
I thank Monika Gosh for English correction.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Lerat, E. Identifying repeats and transposable elements in sequenced genomes: how to find your way through the dense forest of programs. Heredity 104, 520–533 (2010). https://doi.org/10.1038/hdy.2009.165
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2009.165
Keywords
This article is cited by
-
Comparative analysis of transposable elements provides insights into genome evolution in the genus Camelus
BMC Genomics (2021)
-
Producing Genomic Sequences after Genome Scaffolding with Ambiguous Paths: Complexity, Approximation and Lower Bounds
Algorithmica (2021)
-
Repetitive DNA profile of the amphibian mitogenome
BMC Bioinformatics (2020)
-
Diverse mobilome of Dichotomius (Luederwaldtinia) schiffleri (Coleoptera: Scarabaeidae) reveals long-range horizontal transfer events of DNA transposons
Molecular Genetics and Genomics (2020)
-
The landscape of transposable elements and satellite DNAs in the genome of a dioecious plant spinach (Spinacia oleracea L.)
Mobile DNA (2019)
