
Abstract
Repetitive DNA sequences playing critical roles in driving evolution, inducing variation, and regulating gene expression. In this review, we summarized the definition, arrangement, and structural characteristics of repeats. Besides, we introduced diverse biological functions of repeats and reviewed existing methods for automatic repeat detection, classification, and masking. Finally, we analyzed the type, structure, and regulation of repeats in the human genome and their role in the induction of complex diseases. We believe that this review will facilitate a comprehensive understanding of repeats and provide guidance for repeat annotation and in-depth exploration of its association with human diseases.
Similar content being viewed by others

Introduction
Repetitive DNA sequences (repeats) are patterns of nucleic acids that occur in multiple copies throughout the genome1. Both eukaryotic and prokaryotic organisms contain a certain proportion of repeats in the genome2,3,4, particularly mammalians, in which repeats account for 25–50% of their entire genome (Supplementary Fig. S1). For instance, about 50% of the human genome consists of repeats5, while roughly 4% of human genes harbor transposable elements in their protein-coding regions6. Because many of these repeats (~89.5%) are located within introns, they have been erroneously assumed to be non-functional7. However, increasing research indicates the significant impacts that repeats in coding and noncoding regions can have on evolution, gene expression regulation, and variation induction8,9,10. For example, when repeats are present in the coding region they get translated canonically. Not only can non-coding repeats be translated by a non-canonical mechanism11, but even the telomeric repeat RNAs can get translated12. Moreover, recent studies have shown that such repeats are closely related to a variety of diseases, such as genetic disorders (e.g., Hemophilia), neurological diseases (e.g., poly-Q diseases), and cancers (e.g., endometrial, stomach and colorectal cancers)13,14,15. A glossary table (Supplementary Table S1) used to explain acronyms/terminologies in this study is shown in Supplementary Note 1.
DNA sequences can be categorized into three groups according to their recurrence frequency16, as shown in Fig. 1(a). The first group is composed of high-frequency repeats, also known as satellite DNA sequences (satDNAs), which are found in various regions of the chromosomes, including pericentromeric, subtelomeric, and interstitial regions. These sequences typically form constitutive blocks of heterochromatin that are essential components of structures such as centromeres and telomeres17. The length of satDNA repeating units can vary from a few base pairs to over 1 kilobase pairs, forming arrays that can span up to 100 megabases and be repeated over 106 times, making up ~8–10% of the human genome18.

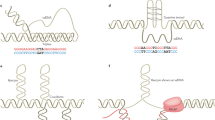
Sub-graph (a): Classification of repeats in the human genome. Sub-graph (b): Arrangement and characterization of repeats in the human genome. Sub-graph (c): Typical structure of DNA transposons, in which TIR and TSD respectively represent the terminal inverted repeat and target site duplication. Sub-graph (d): Typical structure of non-LTR retransposons, in which the color blocks represent the protein domains contained in each family, and the gray block represents the non-coding regions. Sub-graph (e): Typical structure of retrovirus-like LTR retrotransposons, in which LTR represents the long terminal repeat. Sub-graph (f): Typical structure and distribution of TRs in the human genome. Sub-graph (g): Proportion of TRs and active TEs in the human genome. Specifically, LINE-1 and LINE-2 retransposons are represented by L1 and L2 respectively, while SINE-VNTR-Alu retrotransposon and Mammalian-wide interspersed repeats are represented by SVA and MIR. The color arrows represent the repetitive unit (or motif) of each kind of TR, and the light black structure represents the chromosome.
The second group comprises moderate-frequency repeats that are typically 500–300,000 base pairs in length and repeated between 10 and 105 times, accounting for ~30% of all repeats19. These repeats are further classified into two subcategories: (A) microsatellites and minisatellites (VNTR), and (B) dispersed repeats, which are primarily made up of transposable elements (TEs)20. It is worth noting that many moderate-frequency repeats have been implicated in gene expression regulation21.
The third group comprises unique, single-copy DNA sequences, which do not share homology with any other sequences in the genome. Examples of such sequences in the human genome include protein-coding genes (e.g., the globin, ovalbumin, and silk fibroin genes), non-coding RNAs, and regulatory elements that control gene expression22,23. Approximately 40–50% of the total human DNA sequences are single-copy DNA sequences, meaning that about half of the human genome is composed of unique and non-repetitive sequences.
According to the arrangement of repeating units, repeats can be classified into two types: tandem repeats (TRs) and interspersed repeats24, as depicted in Fig. 1(b). Interspersed repeats, also known as transposons or TEs, consist of DNA and RNA transposons25. Generally, TRs refer to a sequence array formed by the repeated occurrence of basic repeating units connected head-to-tail26 (Supplementary Note 2). TRs, especially satellite DNA, are clustered in specific chromosomal regions such as centromeres, tetramers, and telomeres, which play an essential role in cellular processes, including chromosome segregation, genome organization, and chromosome end protection27. For example, centromeres contain long tandem arrays of alpha-satellite repeats that extend over millions of base pairs and are organized in a hierarchical manner. The tandem arrays span between 100 and 5000 bp on different chromosomes, ranging from 0.2 to 10 Mb. Some of these arrays include 17 bp binding motifs for the centromere-specific DNA binding protein, which have been used to create synthetic human chromosomes28.
Tandem repeats
Tandem Repeats in the human genome can be divided into the following subcategories: microsatellites, minisatellites, centromeric satellites, and telomeric and subtelomeric repeats (Fig. 1(f) and Table 1). The difference between microsatellites and minisatellites is represented in their length and frequency of occurrence. Microsatellites are DNA sequences of <5 bp units repeated in tandem and are most frequent in the human genome29. Minisatellites are tandem repetitions of more than 5 bp units, and their frequency in the human genome is relatively rarer than that of the former30. In the human genome, centromeric satellites can be classified into the alpha-satellite and Satellite II/III. Among them, Satellite II/III comprises of various variations on the ATTCC motif31. Telomeric repeats (satellites) are located at the telomeres, consisting of 300–8000 precise CCCTAA/TTAGGG motifs and covering a range of 2–50 kb on the end of the chromosomes32. Subtelomeric repeats are located in the boundary of 100–300 kb between the telomere and the remaining part of the chromosome, consisting of satellite-like sequences33. Type, length, frequency, and distribution of TRs in the human genome are summarized in Table 1 and Supplementary Table S2.
Transposons
Transposons are classified into RNA and DNA transposons, depending on their mode of transposition. RNA transposons use a cut-and-paste mechanism, where the transposase enzyme excises the transposon from its original location and inserts it elsewhere in the genome via an RNA intermediate. DNA transposons also use a cut-and-paste mechanism, but they move directly as DNA and are excised from their donor locus and reinserted elsewhere in a conservative mechanism. This divergence results in various dissimilarities in their transposition mechanisms and evolutionary trajectories. Typical structures of retrotransposons, transposons, and tandem repeats are illustrated in Supplementary Fig. S2(a),(b) and (c), respectively.
DNA transposons, also known as Class II transposons, can be classified into four super families based on their constituent structures and transposition patterns: miniature inverted-repeat TEs (MITEs), Cryptons, Mavericks (or Polintons), and Helitrons. MITEs are non-autonomous transposons primarily found in the non-coding regions of plant and animal genomes34, with the ability to alter gene structures and functions. Cryptons are a unique class of DNA transposons that use Tyrosine Recombinase (YR) to cut and reattach recombining DNA molecules35, allowing them to incorporate YR sequences and drive animal evolution. Mavericks are large DNA transposons commonly found in eukaryotic genomes, with 6 bp target site duplication (TSD) sequences and genes homologous to viral proteins36. Helitrons are recently discovered eukaryotic transposons present in many plant and animal species37, which propagate through a rolling circle mechanism but don’t generate terminal repeats or TSDs. DNA transposons are characterized by terminal inverted repeat sequences (TIRs), which are complementary to each other at the left and right ends of the transposon. These transposons, also known as jumping genes, can move and integrate into diverse genomic regions. Figure 1 (c) illustrates the general structure of DNA transposons in genomes. DNA transposons, which make up about 5% of the human genome38, are considered DNA fossils because no family of them currently remains active in most mammals, including humans39,40.
RNA transposons, also known as retrotransposons or Class I transposons, can be classified into five super families based on their structures and transposition patterns: Long terminal repeats (LTRs), Long interspersed nuclear elements (LINEs), Short interspersed nuclear elements (SINEs), Dictyostelium intermediate repeat sequence (DIRS), and Penelope-like elements (PLEs)41,42. LTR retrotransposons are related to retroviruses and have LTRs at their 5′ and 3′ ends, which likely originated from ancient retroviral infections43. LINEs contain an internal promoter that drives the expression of transposition machinery, including reverse transcriptase and an endonuclease44. SINEs depend on LINEs for their transposition, with specificity determined by their 5′ tails. Most SINEs are derived from tRNA, 7SL RNA, or 5s RNA and have an RNA-Pol III promoter45,46. DIRS retrotransposons, which have tyrosine recombinase, differ from integrases or endonucleases commonly used by retrotransposons for site-specific genomic integration47,48. PLEs share an ancestor with telomerase reverse transcriptases (TERTs) and have unique features in retroelement phylogeny49. In the phylogeny of reverse transcriptases (RTs), PLEs do not belong to the LTR or non-LTR retrotransposon groups but form a sister clade with TERTs. TERTs are major components of the telomerase complex that maintain the linear chromosome ends in most eukaryotes50,51.
The RNA transposons in the human genome can be classified into LTR and Non-LTR retrotransposons. Non-LTR retrotransposons lack LTRs, but contain genes for reverse transcriptases, RNA-binding proteins, nucleases, and sometimes the Ribonuclease H domain52. LINE and SINE are two remaining active super families contained in non-LTR retrotransposons of the human genome, consisting of LINE1 (L1), Alu, and SINE-VNTR-Alu (SVA), three active families (Table 2). Many studies have suggested that L1 may contribute to human cancers by mutating specific oncogenes or tumor suppressor genes in somatic cells53. For example, there is evidence that APC tumor suppressor gene failure is caused by the L1 insertions, which may be an important factor in the development of colorectal cancer54. In addition, Alu elements are retrotransposons specifically present in primate genomes that can regulate gene function by providing canonical polyadenylation signals and play a critical role in the primate genomic diversity, causing complex diseases55. For instance, many complex human diseases, such as meningococcal disease, venous thromboembolism, obesity, and breast cancer, are related to the structural variants caused by Alu insertions56. Currently, SVA is more active than high-copy pseudogenes (e.g., processed ribosomal pseudogenes), and SVA insertions may alter gene expression and cause several human diseases57. For example, SVA regulates the expression of related genes whose insertions have been identified as a significant contributor to diseases such as X-linked dystonia-parkinsonism, Neurofibromatosis type 1, and hemophilia B58, through mechanisms, such as loss of function mutation, modulation of splicing, and deletions at the site of insertion. The general structures of non-LTR retrotransposons are presented in Fig. 1(d). The type, family, and length distribution of repeats, as well as a brief introduction to their biological functions, are shown in Supplementary Table S3.
The general structure of retroviruses and LTR retrotransposons are similar59. Several LTR retrotransposons have similar open reading frames (ORFs) to those of retroviruses, consisting of the gag and pol (pro) genes and, in some cases, env and other accessory genes. The main difference between retroviruses and LTR is the presence of a functional envelope (env) gene in retroviruses, which is absent or nonfunctional in LTRretrotransposons60. The general structure of the retrovirus-LTR is illustrated in Fig. 1 (e). No retrotransposable LTR retrotransposons have been identified in the human genome, and no LTR retrotransposon insertions have been collected in the database of human mutations. However, many elements belonging to the young human endogenous retroviruses (HERV) family, such as HERV-K (K denotes a lysine-tRNA-specific primer binding site to initiate reverse transcription), have an individual ORF domain in their structure capable of translation and production of functional proteins61. Furthermore, HERVs and mammalian apparent LTR retrotransposons (MaLRs) are remnants of ancient retroviral infections found within the human genome. These genetic components are notable for their up-regulation after innate immune activation and are primarily regulated in the context of immunity (Table 2). Retroelements and isolated LTRs, as part of molecular evolution, may benefit the host by promoting plasticity and gene expression regulation (i.e., via promoters and cis-regulatory sequences)62. The expression of HERV-K envelope transcripts is typically undetectable in normal human breast tissues but is detectable in most breast cancer tissues63. Therefore, this expression pattern can be used as a new disease biomarker in clinical diagnosis. The general structure and distribution of tandem repeats, and the percentage of TE families in the human genome are illustrated in Fig. 1(f) and (g), respectively. The proportion of the most abundant repeats in the genomes of Humans, Rice and Drosophila is presented in Supplementary Fig. S1.
Sequence analysis techniques such as de novo assembly, multiple sequence alignment (MSA), sequencing error correction, SNP and variation detection are often impacted by repeats64,65. For example, they are a primary cause of assembly errors in contigs generated by de novo assembly66. Repeats also introduce ambiguity in MSA of sequencing reads, which can interfere with downstream sequencing error correction, SNP identification, variant detection, and gene expression abundance analysis67,68.
Ambiguous paths in assembly graphs such as de Bruijn, string, and overlap graphs are often caused by repeats. Repeats eventually form misassemblies and gaps in contigs, affecting the accuracy and completeness of assemblies and limiting downstream applications (Supplementary Fig. S3(a) and (b))69. Obtaining accurate sequence composition of highly complex short TRs (STRs) in regions such as telomeres, subtelomeres, and centrioles through de novo assembly is challenging70. This limitation severely restricts the study of these regions. Repeats also pose a significant challenge to multiple sequence alignment (MSA), complicating alignment position determination and reducing the performance of sequencing error correction and the sensitivity of detecting SNPs, indels, and other mutations (Supplementary Fig. S3(c))71. A summary of the challenges posed by repeats for sequence analysis is provided in Supplementary Note 3.
Biological functions of repeats and their roles in the human genome
Repeats play crucial roles in biological processes with both functional and non-functional implications. Certain repeats, like promoter and enhancer repeats, regulate gene expression by acting as binding sites for regulatory proteins. They also serve as structural elements, such as centromeres and telomeres, which are vital for genome stability and cell division. Moreover, repeats drive genome evolution through duplication, recombination, and transposition processes. Most repeats in the human genome are derived from TEs, which can move within the genome and act as regulatory elements controlling gene transcription, splicing, and genome architecture, potentially causing mutations or altering genome size and structure72 (Supplementary Fig. S4). In addition, TRs can alter the chromatin structure and affect transcription, leading to gene expression and protein abundance changes, although they represent only a tiny fraction (e.g., TRs accounted for only ~ 3%, as shown in Fig. 1(g)) of the human genome (Supplementary Fig. S5). The biological functions of repeats and their roles in the human genome are discussed in the following sections, and several typical examples of their influence are summarized in Supplementary Note 4.
Biological functions of transposable elements
The movement of TEs may result in mutations, alter gene expression, induce chromosome rearrangements, and enlarge genome sizes due to increased copy numbers73. Thus, they are considered an essential contributor to gene and genome evolution74. In addition, TEs have also been recognized as promising candidates for stimulating gene adaptation through their ability to regulate the expression levels of nearby genes75. Furthermore, combined with their mobility, TEs can relocate adjacent to their targeted genes and control the expression levels of those genes, depending on the circumstances76. The illustrations in Fig. 2 and Supplementary Fig. S4 show how the genome can be affected by TEs in direct or indirect ways.

TEs can directly or indirectly affect the genome through some specific mechanisms.
Transposable elements can cause mutations and genetic polymorphisms
Many TE families are still active and undergoing constant transposition. Variations are induced when TEs transpose nearby genes and regulatory regions, and these are often rare mutations under purifying selection. For example, an experimental study revealed that the spontaneous insertion of multiple TEs causes more than 50% of all known phenotypic mutants in D. melanogaster77. Another experimental study found that ~10–15% of inherited mutant phenotypes in the mouse genome are caused by the autonomous activity of a family of persistently active LTR retransposons8. Furthermore, in another study78, the researchers found that the average difference between any two human haploid genomes is caused by ~1000 TE-dominated insertions, primarily from the L1 or Alu families. The primary mechanisms by which TEs cause mutations and genetic polymorphisms are described subsequently:
Insertion
TEs can insert themselves into new genomic locations, which can result in various types of mutations79. When TEs insert into protein-coding regions, they can disrupt the reading frame, introduce premature stop codons, or alter splicing patterns, leading to loss-of-function mutations. Insertion into regulatory regions can disrupt the binding sites of transcription factors or other regulatory elements, affecting gene expression levels or patterns. These insertional mutations can result in genetic variations and contribute to phenotypic diversity.
Retrotransposition
Retrotransposons, a type of TE, can undergo retrotransposition, where they are transcribed into RNA and then reverse transcribed back into DNA, leading to reintegration at a new genomic location. This process can result in the duplication of TEs and adjacent genomic sequences, creating copy number variations80. Retrotransposition can also lead to the formation of processed pseudogenes, which are nonfunctional copies of genes81. The repeated retrotransposition events of TEs can generate genetic polymorphisms and contribute to the evolution of genomes.
In the human genome, gene mutations and the formation of malignant tumors may be caused by active TEs transposition (Supplementary Note 4). For example, LINEs are a group of non-LTR retrotransposons and are widespread in the genome of many eukaryotes. L1 is the only abundant and active LINE in the human genome, and the human genome contains an estimated 100,000 truncated and 4000 full-length L1 elements accounting for about 17% of the entire genome82. Since L1 correlations with disease and immunity by producing gene mutations, it has become a significant hallmark of several cancers (e.g., ovarian, endometrial, breast, colon, kidney, etc.) and other disorders (Supplementary Table S4). The associations between L1 and some complex diseases and its regulatory mechanism are presented in Fig. 3. In addition, L1 promotes the occurrence of malignant tumors through three main mechanisms: hypomethylation, aberrant integrations, and high expression of its internal ORF1 and ORF2 domains83,84. The relationship between L1 and gene mutations producing malignant tumors is introduced in Supplementary Note 4. Another well-known example is the Alu element, a type of SINE, which can disrupt gene regulation and contribute to genomic diversity and disease susceptibility85. Furthermore, one study reported an association between SVA insertions and neurological diseases such as Parkinson’s disease and amyotrophic lateral sclerosis86. In addition, a recent research has indicated that HERV-KHML-2 insertions can contribute to somatic mosaicism and influence gene expression in certain tissues, potentially impacting disease development87.

For example, hypomethylation, aberrant integration, and highly expressed ORF1 and ORF2 domains of L1 are related to cancers and thus serve as markers for cancer diagnosis.
Transposable elements can regulate gene expression and activity repression
The TE transposition is an essential factor in gene expression variation, often resulting in extreme gene expression changes much more significantly than those produced by rare SNPs88. Involvement in gene expression regulation is another crucial function of TEs in the human genome. There are two primary mechanisms by which TEs regulate gene expression. First, they provide cis-regulatory sequences in the genome with intrinsic regulatory properties for their expression, making them potential regulators of host gene expression. Second, TEs can encode regulatory RNAs. A growing number of studies have demonstrated that their sequences are found in most miRNAs and long noncoding RNAs (lncRNAs), implying that these RNAs are derived from TEs89. Moreover, TEs can be activated or repressed under stress conditions. In some cases, the repression of TEs occurs after the initial activation90. For instance, to suppress TEs activity, host cells have developed a variety of mechanisms, including epigenetic pathways, such as DNA methylation and histone modifications. The primary mechanisms by which TEs regulate gene expression and activity repression are described subsequently:
Epigenetic modification
TEs can influence gene expression by modifying the epigenetic landscape of the genome. TEs often contain regulatory sequences, such as promoters and enhancers, that can interact with nearby genes. The presence of TEs can attract epigenetic modifiers, resulting in the deposition of repressive chromatin marks, such as DNA methylation and histone modifications. These epigenetic modifications can lead to gene repression or silencing by preventing the binding of transcription factors and the access of transcriptional machinery to gene regulatory regions. Conversely, some TEs may also act as regulatory elements, promoting gene activation when demethylated or associated with activating chromatin marks.
Production of non-coding RNAs
TEs can generate non-coding RNAs, such as long non-coding RNAs (lncRNAs) and small interfering RNAs (siRNAs), that play a role in gene regulation. TEs can serve as transcriptional starting sites for the production of lncRNAs, which can interact with chromatin and modulate gene expression. In addition, TEs can be transcribed into siRNAs, which can then guide RNA-induced gene silencing complexes to complementary mRNA sequences, leading to the degradation or repression of target transcripts.
In the human genome, more than 60% of SVAs are within genes or located in their 10 kb flanking regions57. Moreover, SVAs could recruit transcription factors and influence the local chromatin structure, regulating the transcription and expression of nearby genes, as has been demonstrated for human endogenous retroviruses, causing a region to become either accessible or inaccessible to transcriptional machinery. Specifically, how it is regulated depends on the epigenetic marks spread throughout the element91. As described in the previous chapters, the hypomethylation of retrotransposable elements has become an epigenetic mark of several diseases (Supplementary Note 5), such as cancers (Supplementary Fig. S6(a),(b) and (c)). As demonstrated by the regulatory role of L1s in cancer, and changes in epigenetic marks of SVAs, such elements are inappropriately reactivated, possibly leading to the dysregulation of neighboring genes and their associated pathways (Supplementary Fig. S7(a)). For example, a recent study highlighted that certain SVA insertions can act as enhancers and influence the expression of nearby genes in a tissue-specific manner92. Another recent study have shown that Alu elements can act as enhancers or repressors and contribute to tissue-specific gene regulation93. The relationship between SVAs and gene expression regulation is presented in Supplementary Note 5.
Transposable elements can associate with genome rearrangement
In reality, TEs can be associated with genome rearrangement through various mechanisms, such as de novo TE insertion, TE insertion-mediated deletion, and homologous recombination between them. These rearrangements increase the genomic difference between genomes, and some specific rearrangements may lead to complex diseases94. As an illustration, the expression of retrotransposition-competent TEs may result in additional insertions, which may affect the expression or function of genes95 and trigger chromosome rearrangements through an ectopic recombination between repeated copies of a TE, causing mutations96, resulting in several complex diseases, such as cancers97, Alzheimer’s disease98, and autoimmune and neurological disorders99. The primary mechanisms by which TEs associate with genome rearrangement are described subsequently:
Transposition
TEs are mobile genetic elements that can undergo transposition, a process in which they move from one genomic location to another. During transposition, TEs can insert themselves into new sites within the genome, leading to rearrangements. For example, when TEs transpose and insert themselves between genes, they can disrupt gene order, create gene duplications, or cause gene deletions. These structural changes can have significant effects on the organization and function of the genome.
Recombination
TEs can serve as recombination sites in the genome, promoting genomic rearrangements. In some cases, recombination events between different TEs or between TEs and their target sequences can result in large-scale genomic rearrangements. This includes chromosomal inversions, translocations, and deletions, which can alter gene order, disrupt regulatory elements, and impact the overall genomic architecture.
Compared to other TEs, Alu and L1 elements in the human genome are more likely to cause genomic rearrangements due to their widespread presence. Specifically, 492 Alu recombination-mediated deletions (ARMDs) have been identified in the human genome, deleting ~400 kb of human genomic sequences, including exons of known or predicted genes100. The ARMD process has significantly contributed to genomic and phenotypic variations between humans and chimpanzees since their evolutionary divergence. For another example, a recent research suggests that L1 insertions can cause genomic rearrangements, including deletions, inversions, and duplications, leading to structural variations in the human genome101. The specific relationship between genome rearrangements caused by TEs and complex diseases is discussed in Supplementary Note 6.
Transposable elements can act as insertional mutagens in germline and somatic cells
Mobile elements, such as L1, Alu, SVA and HERV-K, are in charge of novel germline insertions, which may lead to genetic illness (Table 3) (Supplementary Note 6.1 to Note 6.7). The primary mechanisms by which TEs act as insertional mutagens in germline and somatic cells are described subsequently:
Disruption of coding sequences
When a TE inserts within a coding region of a gene, it can disrupt the reading frame, introduce premature stop codons, or cause other structural changes. This disruption can lead to the loss of gene function or the production of truncated and non-functional proteins. In germline cells, such mutations can be inherited and contribute to genetic variation in subsequent generations.
Alteration of regulatory elements
TEs can insert near regulatory elements, such as promoters, enhancers, or insulators, and disrupt their function. This can result in the misregulation or aberrant expression of genes. Changes in the regulation of critical genes can have profound effects on cellular processes, development, and disease susceptibility.
For instance, a study has revealed that over 120 independent TE insertions are essential contributors to human diseases, including hemophilia, Dent disease, neurofibromatosis and cancers102. The germline transposition rate for the Alu element in humans is about 1 in 21 births103, while the corresponding value for the L1 element is about 1 in 95 births104. Historically, TEs have generally been considered transcriptional silencing in somatic cells. However, evidence indicates that active TEs are also present in the somatic cells of various organisms. As an illustration, the expression and transposition of the L1 element have been identified in several somatic contexts, such as early embryos and specific stem cells105. Furthermore, HERV-K elements have been implicated in insertional mutagenesis. Recent studies have identified HERV-K insertions with potential mutagenic effects on nearby genes, including cancer-related genes106 (Supplementary Fig. S7(b)). Human cancers have also exhibited somatic activity, with tumors able to pick up hundreds of additional L1 insertions. For instance, recent research has highlighted the impact of L1 insertions in diseases such as cancer, neurological disorders, and genetic syndromes107.
Transposable elements can drive key coding and non-coding RNAs
According to mounting evidence, TE insertions may serve as the building blocks for forming protein-coding genes and non-coding RNAs that can carry out the crucial physiological functions of cells108. For example, Rag1 and Rag2 are spectacular examples of deeply conserved TE-derived genes that activate V(D)J somatic recombination in the immune system of vertebrates109. As another example, based on a mixed lncRNA annotation from RNA sequencing and GENCODE (a scientific project in genome research and part of the ENCODE scale-up project), a study estimated that 41% of lncRNA nucleotides are derived from TEs, and the majority of lncRNAs (about 83%) contain at least one TE fragment110. The primary mechanisms by which TEs drive key coding and non-coding RNAs are described subsequently:
Retrotransposition
TEs, particularly retrotransposons, can undergo a process called retrotransposition where they are transcribed into RNA and then reverse transcribed back into DNA, leading to their insertion into new genomic locations. If these retrotransposed elements land within or near functional genes, they can act as alternative promoters, enhancers, or splice sites, giving rise to new coding and non-coding RNA transcripts. This process can generate novel RNA molecules with potentially functional roles in cellular processes.
Co-option of regulatory elements
TEs often contain regulatory sequences such as promoters, enhancers, and insulators. These sequences can be co-opted by the host genome to regulate the expression of nearby genes or to shape the expression patterns of non-coding RNAs. By providing alternative regulatory elements, TEs can impact gene expression networks and contribute to the production of key coding and non-coding RNAs.
The presence of TEs that drive key coding and noncoding RNAs in the human genome may be associated with certain diseases (Table 3). For instance, HERVs affect human health and cause disease by encoding proteins, acting as promoters/enhancers or lncRNAs, accounting for about 9% of the human genome111. HERVs can also have a direct effect via their proteins in the development of cancers. For example, by inducing cell-cell fusion or epithelial-to-mesenchymal transition, HERV envelope proteins play a critical role in tumorigenesis and development in melanoma, endometrial carcinoma, and breast cancer112. Furthermore, HERVs can generate lncRNAs that promote cancer proliferation, motility, and invasion. For example, in the study113, researchers have found that several HERVs-derived lncRNAs, such as UCA1, SAMSON, and BANCR, are involved in the processes of proliferation, motility, and invasion in bladder cancer and melanoma. The relationship between transcriptional activation of HERV retrotransposons and human cancer is summarized in Supplementary Note 6.7.
Transposable elements can alter transcriptional networks and conduce to cis-regulatory DNA elements
Cis-regulatory DNA elements (CREs) are regions of non-coding DNA that regulate the transcription of neighboring genes. In addition, CREs are vital components of genetic regulatory networks. Some TEs have evolved into CREs, whose function is to mimic host promoters, enabling them to recruit host-encoded factors driving their selfish transcription114. For instance, due to innate and adaptive immune responses, the immune system can protect organisms from pathogens and foreign substances. During evolution, some TE families, including many endogenous retroviruses (ERVs), have the capacity to influence and shape transcriptional networks. They can function as signaling molecules that regulate DNA elements and the immune system115. The primary mechanisms by which TEs alter transcriptional networks and conduce to cis-regulatory DNA elements are described subsequently:
Enhancer hijacking
TEs can integrate near enhancer regions, affecting the binding of transcription factors and changing the regulation of nearby genes.
Promoter modulation
TEs can also insert near gene promoters, influencing the recruitment of transcriptional machinery and impacting gene expression levels.
In the human genome, L1 elements have the potential to influence transcriptional networks. Recent research has demonstrated that L1 retrotransposition can introduce novel regulatory elements, alter gene expression patterns, and contribute to cellular diversity116. Furthermore, Alu elements can also impact transcriptional networks. Recent studies have highlighted their role in shaping tissue-specific gene expression, alternative splicing, and influencing the expression of neighboring genes through enhancer or promoter activities117. The diverse mechanisms through which TEs influence host gene-regulatory networks can be broadly categorized into five classes: (1) introduction of transcription factor binding sites, promoters, and enhancers, (2) modification of 3D chromatin architecture, (3) production of regulatory non-coding RNAs, (4) usage of TE-derived coding sequences as new transcriptional effector proteins, and (5) secondary effects of TE silencing mechanisms118.
Biological functions of tandem repeats
TRs are common features of both prokaryote and eukaryote genomes. For example, more than one million distinct TRs are contained in the human genome, many of which are highly polymorphic in sequence composition and copy number. TRs can be found in intergenic regions and in both the non-coding and coding regions of a variety of genes119,120,121. Moreover, TRs occur near or between a series of genes and can affect the structure and function of DNA, RNA, and proteins through specific mechanisms and produce a series of molecular and cellular consequences122. As an illustration, many TRs are involved in biological functions in a copy number-dependent manner, and there is evidence that TRs may regulate the expression of nearby genes by altering their copy number123. In general, TRs are highly mutable and can be located in exons, introns, or intergenic regions, providing opportunities for the modulation of gene expression, as well as the structure and function of RNAs and proteins124. Expanded TRs usually cause various disorders, including autism spectrum disorder (ASD) and cancers (Table 3 and Supplementary Table S5). The illustrations in Fig. 4 and Supplementary Fig. S5 highlight how TR can directly or indirectly affect the genome.

Similar to TEs, TRs can also affect the genome in specific ways.
Tandem repeats can accelerate evolution and adaptation
TRs are often referred to as satellite DNA, which can be further classified into microsatellites or short tandem repeats (STRs) (motif length: 1–4 bp), minisatellites (motif length: 5–64 bp), and macrosatellites (motif length: several kp), according to the size of the repeated motifs125. For example, slipped strand mispairing is a mutation process that occurs during DNA replication, which is one explanation for the origin and evolution of repetitive DNA sequences126. TRs, especially STRs, are extremely unstable in terms of length, sequence composition, and copy number, with mutation rates typically 10–100,000 times higher than in other parts of the genome127. These unstable repeats are found in up to 20% of eukaryotic genes and promoters, where they confer phenotypic or functional variability on the cell surface and extracellular proteins and have pathological consequences. The primary mechanisms by which TRs accelerate evolution and adaptation are described subsequently:
Rapid genetic variation
TRs undergo rapid changes in copy numbers and lengths, creating genetic diversity that can drive the emergence of new traits.
Gene regulation
TRs located in regulatory regions can influence gene expression, allowing for adaptive changes to occur in response to environmental pressures.
In the human genome, TRs are also frequently found in genes that control body morphology128,129. For example, compared with synteny blocks, evolutionary breakpoint regions in the human genome contain more base pairs associated with TRs, with AAAT being the most frequent motif130. These TRs within evolutionary breakpoint regions have the potential to facilitate and accelerate gene expression evolution and generate sufficient variability to drive the rapid evolution and adaptation of organisms131. Furthermore, recent studies have shown that STR variations in immune genes, such as HLA loci, can shape immune responses and contribute to adaptation to diverse environments132. In addition, TRs located in regulatory regions can facilitate evolutionary adaptations. Recent research has suggested that expansion or contraction of STRs within regulatory regions can modulate gene expression and contribute to phenotypic variation and adaptive responses133.
Tandem repeats can play a critical role in the structural stability of genetic materials during the cell cycle
Within or around certain specialized chromosomal regions (e.g., centromeres, telomeres, and subtelomeres), TRs may play crucial roles in the structural stability of genetic materials during the cell cycle134. The primary mechanisms by which TRs play a critical role in the structural stability of genetic materials during the cell cycle are described subsequently:
Replication fork stabilization
TRs, consisting of repeated DNA sequences adjacent to each other, can stabilize the replication forks during DNA replication. The repetitive nature of TRs provides a stable template for DNA polymerases to bind and initiate replication. This stability prevents replication forks from stalling or collapsing, ensuring accurate and complete DNA replication. TRs act as essential structural elements that contribute to the stability of genomic regions during the cell cycle.
Telomere maintenance
Telomeres, specialized TRs located at the ends of chromosomes, play a crucial role in maintaining genomic stability. Telomeres protect the ends of chromosomes from degradation, fusion, and recognition as DNA breaks. During each round of DNA replication, the conventional DNA replication machinery has difficulty fully replicating the ends of linear chromosomes. Telomeres, with their repeated sequences and associated proteins, form a protective cap that allows complete replication of chromosome ends and prevents the loss of genetic information. Telomeric TRs, in conjunction with telomerase enzyme activity, ensure the integrity and stability of the genome during successive cell divisions.
For instance, centromeres are chromosomal domains responsible for the faithful transmission of genetic material during cell division. They are characterized by highly repetitive DNA regions and bound kinetochore proteins, and they are required for the attachment of microtubules to the chromosomes during mitosis135. An array of tandem repeats known as alpha-satellites is one of the crucial components of centromeres, and it plays a vital role in maintaining the stability of human chromosomes. Variations in alpha-satellites can impact the function of the centromere136. In addition, telomeres consist of repeat sequences and are bound by multiple telomeric interacting proteins. In mammalian cells, telomere DNA is composed of double-stranded tandem repeats of TTAGGG, with terminal 3′ G-rich single-stranded overhangs. Telomeres are protected by protein complexes, such as shelterin, which includes TRF1, TRF2, POT1, and other proteins that interact with telomeres indirectly137. This protection distinguishes natural chromosome ends from accidental DNA breaks and prevents unwanted repair machinery activity on telomeres.
Furthermore, the 5′ and 3′ UTRs of genes are transcribed but usually not translated into proteins. However, they contain various regulatory elements involved in post-transcriptional gene regulation, such as mRNA stability, localization, and translation efficiency138. STRs within UTRs can contribute to gene regulation in the following ways: (1) Modulation of mRNA stability: STRs in the UTRs can impact the stability of mRNA molecules. Changes in STR length may affect the folding of UTRs, leading to altered interactions with RNA-binding proteins and subsequent degradation or stabilization of mRNA. (2) Regulation of translation efficiency: UTRs can also influence translation initiation and efficiency. STRs located in the 5′ UTRs can affect ribosome binding and start codon recognition, leading to changes in translation rates and protein production. STR variations in UTRs have been associated with complex traits and diseases. For instance, a recent study identified UTR STR expansions associated with the risk of neurodevelopmental disorders139.
In addition, TRs can be transcribed into RNA molecules through the process of transcription, which is carried out by RNA polymerases140. When these TRs are transcribed into RNA, the resulting RNA molecules can exhibit structural features and functional implications. The structure of TRs in terms of transcribed RNA are as follows: (1) Transcribed RNA molecules derived from TRs retain the repetitive nature of the underlying DNA sequence. (2) TR RNA can fold into various secondary structures due to intra-molecular base pairing within the repetitive sequence. (3) TR-derived RNA molecules can serve diverse non-coding RNA functions. For example, some TR RNAs act as scaffolds for the assembly of ribonucleoprotein complexes or regulate gene expression through interactions with RNA-binding proteins or microRNAs. (4) TR-derived RNA can engage in regulatory mechanisms such as RNA interference, where complementary TR RNA pairs with target mRNA to modulate its stability or translation. TR RNA molecules can also influence cellular processes by sequestering RNA-binding proteins or acting as decoys for regulatory factors. (5) Expansions or contractions of TRs in transcribed RNA have been linked to various genetic diseases. Abnormal TR RNA structures and interactions can result in functional consequences, including the sequestration of RNA-binding proteins, disruption of cellular processes, or induction of toxic effects. These factors contribute to the pathogenesis of diseases141,142.
Tandem repeats can result in redundancy of gene families and functions
A gene family is a collection of many related genes that typically perform comparable biological tasks. Individual members of clustered gene families are often responsible for achieving specific phenotypes or functions in the overall mission143. Tandem gene duplication is thought to have significantly contributed to the evolution of large gene families, genetic and morphological diversity, and speciation in eukaryotes144,145. The primary mechanisms by which TRs result in redundancy of gene families and functions are described subsequently:
Gene duplication
TRs can undergo replication slippage during DNA replication, leading to the expansion of the repeat region and subsequent gene duplication. This process can result in the creation of additional copies of genes within the same genomic region. The duplicated genes are often subject to variations, such as point mutations or insertions/deletions, that accumulate over time, leading to divergence in their sequences and functions. This duplication and subsequent diversification of gene copies can result in redundancy within gene families, where multiple genes have similar or overlapping functions.
Divergent evolution
Over time, duplicated genes arising from TRs can undergo divergent evolution. Mutations and genetic changes accumulate in each gene copy, resulting in alterations to their coding sequences and regulatory elements. These changes can lead to functional divergence, where duplicated genes acquire different functions or have differential expression patterns. As a result, redundant gene copies can contribute to the expansion and diversity of gene families, providing evolutionary opportunities for gene innovation and adaptation to new environmental or physiological contexts.
For example, the genes responsible for coding ribosomal RNA (rRNA) are present in the human genome as numerous tandemly arrayed copies. These ribosomal DNA (rDNA) repeats facilitate the production of abundant amounts of rRNA to satisfy the cell’s constant requirement for ribosome production146. In mammals, rDNA repeats are present in two types of tandem arrays, termed the 5S and 47S (or 45S) arrays. The 5S rDNA repeats are located in one large tandem repeat array on chromosome 1 in humans. The 47S arrays are located on the short arms of five acrocentric chromosomes in humans (chr. 13, 14, 15, 21, 22)147. Research conducted by the Chinese Academy of Sciences investigated the impact of TR-mediated expansions and variations within the mucin gene family. These TR expansions and variations contribute to the redundancy and functional diversification of mucins, which play important roles in various cellular processes148.
Tandem repeats can regulate gene expression, and their expansion can cause a range of disorders
TR instabilities, especially microsatellite instability, contribute significantly to causing gene expression variation in humans149, and numerous disorders such as cancer, ASD, Huntington’s disease, various ataxias, motor neuron disease, frontotemporal dementia, and fragile X syndrome, are associated with the expansion of TRs, particularly STRs150,151,152,153,154 (Table 3). The primary mechanisms by which TRs regulate gene expression, and their expansion can cause a range of disorders are described subsequently:
Transcriptional modulation
TRs located within gene regulatory regions, such as promoters and enhancers, can influence gene expression by affecting the binding of transcription factors. The presence of TRs can alter the three-dimensional chromatin structure, leading to changes in the accessibility of regulatory elements and the recruitment of transcriptional machinery. The variability in TR length and sequence can impact the affinity of transcription factors for binding sites, resulting in differential gene expression levels.
Epigenetic regulation
TRs can act as susceptible targets for epigenetic modifications, such as DNA methylation and histone modifications. The length and sequence composition of TRs can influence the degree of epigenetic regulation. Methylation of TRs, for example, can lead to the formation of repressive chromatin and transcriptional silencing. These epigenetic modifications can have a profound impact on gene expression patterns and contribute to the regulation of various cellular processes.
Alternative splicing
TRs within exons or introns can affect alternative splicing, a process that generates multiple mRNA isoforms from a single gene. Variation in TR length can influence the splicing process by altering the stability of RNA secondary structures or serving as binding sites for splicing factors. This can result in the inclusion or exclusion of specific exons, leading to the production of different protein isoforms with distinct functions or regulatory properties.
Expansion
The expansion of TRs can also cause a range of disorders, known as trinucleotide repeat expansion disorders. When the size of certain TRs exceeds a threshold, it can lead to genomic instability and pathological consequences. The expanded TRs can exhibit a tendency for further expansion and accumulation in subsequent generations, resulting in a dynamic and progressive increase in repeat length. The expanded TRs can interfere with gene function, leading to impaired protein production, altered protein structure, or disrupted cellular processes. Trinucleotide repeat expansion disorders include conditions like Huntington’s disease, Fragile X syndrome, and several forms of spinocerebellar ataxia, among others. These disorders often display a correlation between the size of the repetitive expansion and the severity of the disease phenotype.
For example, Lynch syndrome is an autosomal dominant disorder that increases the risk of developing colorectal cancer, endometrial adenocarcinoma, and tumors of the small intestine, stomach, ureter, renal pelvis, ovary, brain, and prostate. Research in study155 has demonstrated that most (90%) colorectal cancer due to Lynch syndrome have microsatellite instability. In addition, researchers in study156 have revealed that one neurodegenerative disease in which microsatellite instability contributes to a substantial number of cases is amyotrophic lateral sclerosis (ALS), a rapidly progressive and uniformly fatal motor neuron disease. Recent research indicates that TR polymorphisms can also regulate gene expression in healthy individuals133. Furthermore, TR instability can lead to reduced gene expression, increased disease incidence, and enhanced tumor aggression (Supplementary Fig. S7(c) and (d)). The association between tandem repeat instabilities and cancer, autism, as well as neurological disorders, is discussed in Supplementary Note 6.8 and Note 6.9.
Repeat detection
Numerous computational methods have been proposed for identifying repeats in genomes, which can be divided into homology-based, structure-based, de novo methods, and hybrid frameworks, as shown in Table 4 and Supplementary Fig. S8.
Homology-based identification methods
Homology-based methods identify repeats by finding subsequences similar to known repeats, which must rely on algorithms for comparing similarity between sequences, such as the hidden markov model (HMM)-based comparison algorithm, and specific databases, such as RepBase157, Dfam158, msRepDB159, REXdb160, and Pfam161. RepeatMasker (https://www.repeatmasker.org) is a representation of such tools, which uses Dfam or RepBase as the backend library and RMBLAST (http://www.repeatmasker.org/RMBlast.html) as the aligner. RMBLAST and Dfam are a new aligner and database specially developed by RepeatMasker team for repeat detection based on the existing aligner BLAST162 (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and database RepBase (https://www.girinst.org/repbase/). Both RMBLAST and Dfam have become gold standards in the field of repeat annotation. Typical homology-based detection methods also include Censor163, TESeeker164, Greedier165, and T-lex166 (Supplementary Table S6). The advantages of homology-based methods lie in their accuracy and the ability to discover families with a small number of copies. Their disadvantage is that they cannot be used to discover new repetitive sequences that are not collected in homology databases. A detailed introduction to homology-based methods can be found in Supplementary Note 7.1.1.
Structure-based identification methods
Repeats, especially TEs, have specific structures, such as the structure of a protein, or non-coding domains, and differ in the presence and size of the TSD, a short, direct repeat generated on both flanks of a TE upon insertion167. Structure-based methods rely on prior knowledge of structural features of known repeats collected in the library and employ a heuristic algorithm to identify repeats in genomes. Typical structure-based identification methods include LTRharvest168, MASiVE169, MGEScan-LTR170, TE-greedy-nester171, SINE-Finder172, SINE_scan173, AnnoSINE174, FINDMITE175, MUST176, detectMITE177, MITE-Hunter34, MITE-Digger178 and, MITE Tracker179 (Supplementary Table S7). The advantages of structure-based methods include high detection efficiency and lower false-positive rate, and the detected repeats are easier to verify and classify. Their disadvantages are that they cannot be used to identify repeats whose structural features are unknown or whose structural features cannot be obtained accurately and completely due to the insufficient precision and completeness of the input sequences. Thus, the detection integrity of such methods is often unsatisfactory. Besides, structure-based methods are often designed for a particular class of transposons (e.g., LTRs, SINEs, and MITEs). Therefore their versatility is limited. A detailed introduction to structure-based detection methods is shown in Supplementary Note 7.1.2.
De novo identification methods
The de novo methods are more flexible than the other two classes of methods because they do not require prior knowledge about the structure or similarity to known repeats180, which can also be classified into three categories based on the core technology that each method depends on. The first class of methods includes Repeat Pattern Toolkit181, RECON182, PILER183, LTRdigest184, and LongRepMarker185, identifying repeats through MSA. The strategy of high-frequency k-mers and space seed extension is used in the second category of methods to identify repeats. The sequences to be detected are converted into k-mers of a certain length, and k-mers whose frequency exceeds a certain threshold are chosen as seeds. Then, the locations of these seeds in the genome are recorded, and the repeats are obtained by performing sequence extensions at both ends of the genome. During the extension process, the detection algorithm always judges whether the extended arrangements are consistent across multiple genome locations. If yes, continue; otherwise, terminate. RepeatFinder186, RepeatScout187, ReAS188, and Generic Repeat Finder (GRF)189 are representative of this class of approaches. The third class of methods includes RepARK190, REPdenovo191, RepAHR192, and RepLong193, which rely on de novo sequence assembly and community detection in sequence similarity network to identify repeats (Supplementary Table S8). Among these four tools, the first three obtain repeats by performing assembly of high-frequency reads or k-mers (Supplementary Fig. S9(a),(b),(c),(d) and (e)). The last method constructs the similarity network by getting the overlaps between long reads, and then use the community discovery algorithm to get the repeats (Supplementary Fig. S9(f)). A detailed introduction to the de novo identification methods is shown in Supplementary Note 7.1.3.
Tandem repeat and their expansion identification methods
Several tools are available for detecting TRs and their expansions, such as mreps194, Tandem Repeats Finder (TRF)195, T-REKS196, TRASH197, EnsembleTR198,199, RExPRT200, GangSTR201, ExpansionHunter200, ExpansionHunter De novo202, Straglr203, and STRling204. Among them, mreps excels by detecting all types of tandem repeats in an entire genomic sequence simultaneously. It incorporates a resolution parameter to identify fuzzy repeats with variations within the repeated units. TRF uses sequence alignment and statistics to detect consecutive repetitive motifs. It gives detailed information about identified repeats, including positions, consensus sequence, length, and alignment scores. This information is valuable for genome analysis, gene mapping, investigating structural variations, and understanding repetitive elements in biology and evolution. T-REKS operates by dividing the input sequence into overlapping k-mer segments, where k is a user-defined parameter. Then, it employs the k-means clustering algorithm to group similar k-mers together, identifying potential TRs. EnsembleTR and GangSTR, developed by the Gymrek Lab, are powerful tools in computational genomics and human genetics. EnsembleTR takes VCF files with TR genotypes for multiple samples and generates a consensus set of genotypes. RExPRT is a machine learning tool used to differentiate pathogenic from benign TR expansions. GangSTR is a tool used for profiling TRs across the genome using short reads. One notable advantage of GangSTR is its ability to handle repeats that exceed the read length. ExpansionHunter and ExpansionHunter De novo are two computational methods developed by Illumina Inc. to locate both known and novel repeat expansions in short-read sequencing data. Straglr is a specialized tool designed to identify and genotype TR expansions using whole genome long-read sequences. STRling is a method for detecting new short TR (STR) expansions from short-read sequencing data, even when no corresponding STR is present in the reference genome.
Hybrid frameworks
The classification of methods mentioned above is based on the core technology utilized in each method. However, there are certain detection tools like Extensive de novo TE Annotator (EDTA)205 and RepeatModeler2206, which employ multiple existing detection algorithms or strategies to perform repeat annotation. These tools cannot be easily classified into the above-mentioned three categories due to their unique approach that incorporates multiple existing methods for repeat annotation. For example, EDTA incorporates various tools, such as RepeatModeler and RepeatMasker, which employ homology-based methods, as well as TransposonPSI. In addition, it incorporates structure-based methods like LTRharvest and LTR_retriever. RepeatModeler2 is another hybrid framework, that utilizes the de novo methods RECON and RepeatScout, along with the Dfam database and the alignment search tool RMBLAST, to identify and model repetitive elements in DNA sequences. Performance comparisons between different repeat detection methods are shown in Supplementary Tables S9–S32 of the Supplementary Note 7.2.
Automated classification and masking of repeats
Classification and masking are two necessary steps after the detection stage in the workflow of repetitive DNA sequence analysis. Precise classification and comprehensive masking of repeats are essential for analyzing their critical roles in genomes. The output of the detection stage consists of raw repeat consensus sequences without any information about the type, structure, and function. The purpose of classification is to classify unknown repeats into their main taxonomic branches (e.g., LTR, LINEs, SINEs, DIRS, PLEs, MITEs, Cryptons, Helitrons, Mavericks, Satellites, low complexity sequences, etc.), and to distinguish their structures and functions. The purpose of repeat masking is to mask the repeats in the genome of a specific sequencing sample with the well-classified elements collected in the repeat database using pairwise sequence alignment algorithms, such as nhmmer, cross_match, AB-BLAST/WU-BLAST, RMBLAST, and Decypher, and to report all locations, specific classifications and copy number information of the hit sequences. The principle of repetitive DNA sequence classification and masking is presented in Fig. 5.

Sub-graph (a): A simple example of the distribution characteristics of repeats in the reference genome, where the black blocks represent chromosomes. Sub-graph (b): Principle of repeat detection, where the final sequence composed of colored bases represents the consensus repeat sequence. Sub-graph (c): Principle of automatic repeat classification, where black and dark green cylinders represent nucleic acid and protein libraries, respectively. Sub-graph (d): Principle of automatic repeat masking. Light green cylinders in Sub-graphs (c) and (d) represent the user-defined repeat library, and black blocks in Sub-graph (d) indicate the sequencing reads from various samples of the same or similar species.
Databases that support automated repeat classification and masking
An accurate and comprehensive repeat database is essential for the automated classification and masking of repeats in genomes. Three well-known nucleic acid libraries, RepBase, Dfam, msRepDB, and three famous protein libraries, RepeatsDB, REXdb, and Pfam, have been proposed to support the automated classification and masking of repeats. RepBase (https://www.girinst.org/repbase/) is a database of prototypic sequences representing repetitive DNA from different eukaryotic species, which currently contains more than 38,000 sequences of different families. Dfam (https://www.dfam.org/releases/Dfam_3.5/) database is an open collection of TEs and genome annotations, which currently houses 285,542 TE models across 595 species and incorporated into the new version of RepeatMasker. msRepDB (https://msrepdb.cbrc.kaust.edu.sa/pages/msRepDB/index.html) is the most comprehensive multi-species repeat database, which currently contains TEs of more than 84,000 species. RepeatsDB (https://repeatsdb.bio.unipd.it/) collects protein structures of annotated TRs, which provides users with the possibility to access and download high-quality datasets either interactively or programmatically through web services. Pfam (http://pfam.xfam.org/) is a database of protein families, which contains many protein families, each of which is represented by MSAs and HMMs. REXdb (http://repeatexplorer.org/?page_id=918) is a reference database of TE protein domains employed in the repeat analysis tools RepeatExplorer217 and DANTE207, which are available on the Galaxy server (https://repeatexplorer-elixir.cerit-sc.cz/). A detailed introduction to repeat databases is shown in Supplementary Note 7.3.2. A performance comparison of the databases is presented in Supplementary Tables S33–S41.
Automated repeat classification methods based on homology searching
The goal of classification is to classify unknown repeats into their main taxonomic branches, which usually refers to the classification of TEs (Fig. 5(a), (b) and (c)). Some methods are proposed based on manually predefined features for automatically classifying TEs, such as TEclass208, RepeatClassifier206, PASTEC209, and REPCLASS210. Homology-based searching and structural features of TEs (e.g., TSD, TRs, tRNA, poly-A signals, SSR, and protein-coding domains) are used in these tools to perform classification (Table 5).
For instance, TEclass (http://www.compgen.uni-muenster.de/teclass) uses support vector machine (SVM) and oligomer frequencies to classify TE consensus repeat sequences into DNA transposons and retrotransposons, including LTRs, LINEs, and SINEs. RepeatClassifier (https://github.com/Dfam-consortium/RepeatModeler) is a homology-based classification module designed in the hybrid TE family discovery framework RepeatModeler2, which compares TE families to RepeatMasker repeat protein databases (e.g., Pfam, REXdb) and RepeatMasker repeat nucleic acid libraries (e.g., RepBase and Dfam) using the homology-based aligner BLAST. PASTEC (http://urgi.versailles.inra.fr/Tools/PASTEClassifier) obtains the similarities and structural features of TEs using profile HMMs211 and homology-based search algorithms (e.g., tblastx, blastx, and blastn) and then classifies TEs into their respective order. REPCLASS (http://sourceforge.net/projects/repclass/) is a tool that automates the classification of TE sequences using control repeat libraries and structural and homology characterization modules, which can classify accurately virtually any known TR types.
Automatic repeat classification methods based on machine and deep learning
Convolutional neural networks (CNNs) are automatic and adaptive representation learning and feature extraction algorithms that can be applied to predict unknown sequence profiles or motifs and functional activity discovery without pre-defining sequence features. Some TE classification algorithms are proposed based on CNNs, among which DeepTE212 and TERL213 are representatives (Table 5).
DeepTE (https://github.com/LiLabAtVT/DeepTE) tra- nsforms sequences into input vectors through a k-mer counting strategy, and classifies TEs into superfamilies and orders based on a tree-structured classification process and eight trained models (class model, classI model, LTR model, nLTR model, SINE model, LINE model, classII_sub1 model and domain model). Among these models, class model is responsible for classifying TEs into Class I, Class II_sub1 and Class II_sub2 transposons, and “ClassI model” is to classify TEs into LTR and non-LTR transposons. Moreover, the false classification correction model and distinction algorithm for distinguishing non-TEs and TEs are also integrated into DeepTE. TERL (https://github.com/muriloHoracio/TERL) is a fast and flexible deep CNN-based approach for classifying TEs and other biological sequences, which employs deep CNNs to preprocess and translate one-dimensional nucleic acid sequences (i.e., image-like data of nucleic acid sequences) into two-dimensional space data. TEclass is an automated classification algorithm based on machine learning support vector machine (SVM). The classification obtained using TEclass is very sparse relative to the overall TE classes, usually only including DNA transposons, LTRs, LINEs, and SINEs. Besides, TEclass can only roughly distinguish non-TE sequences, but cannot accurately classify them. Compared with TEclass, TERL can distinguish non-TE sequences and label numerously of unknown types of repetitive sequences in the detection results as corresponding non-TE types, which greatly improves the accuracy of non-TE sequence identification. In addition, TERL has excellent scalability and can be executed seamlessly in GPUs, greatly improving the efficiency of data processing.
Automated masking of repeats
Repeat masking is also a vital step in the pipeline of genome repeat analysis (Fig. 5(D)). Three steps of detection, classification, and masking are integrated into some hybrid repeat detection frameworks, such as RepeatMasker, RepeatModeler, and LongRepMarker, to obtain classified TEs (e.g., LTRs, LINEs, SINEs, etc.) and masking reports (e.g., the length occupied, coverage ratio, and location of each TE in the genome). As described, RepeatMasker (https://www.repeatmasker.org/) is a robust detection and masking framework based on homology searching. The input of RepeatMasker are the genome to be annotated and a standard repeat library, such as the RepBase or Dfam. During the masking process, RepeatMasker aligns the well-classified TEs collected in the repeat library to the sequences of the genome one by one, records the length occupied, coverage ratio, and location of each TE in the genome, and generates a masking report. Performance analyzes of automated repeat sequence classification and masking methods are shown in Supplementary Tables S42–S46.
Discussion
In this section, we summarize the challenges and solutions in the research field of genomic repeat detection and annotation, as well as future development trends.
Since not requiring prior knowledge, the de novo methods are more flexible and valuable than the homology-based and structure-based methods. However, developing advanced de novo algorithms for comprehensive repetitive DNA sequence detection is challenging due to the short length of NGS reads and the high rate of sequencing errors in TGS (Third-generation sequencing) reads. A hybrid strategy combining short and long reads is currently the most effective way to achieve the above goals. However, before implementing the hybrid strategy, we need to obtain multiple sequencing data, such as NGS reads, TGS reads, and even 10× genomic reads, of the same sample in advance, resulting high detection costs and difficult algorithm design. Therefore, successfully overcoming the impact of sequencing errors in TGS reads and directly carrying out high-precision and ultra-complete repeat detection using the increasing number of high-quality TGS reads will become a research focus in the future. Furthermore, the variation of TRs is closely related to the emergence of complex diseases, such as cancers, neurological disorders, and autism. However, there has not been much progress in the development of algorithms for the detection of TRs and their expansions. Databases containing TRs of multiple species are also very scarce. Therefore, researching superior identification methods for TRs and complete TR databases is of great significance in exploring their biological functions in genomes, which is another important research focus in the future.
Several automatic repeat classification methods have been proposed based on machine and deep Learning. These methods all benefit from SVM and CNNs and perform better than traditional methods in some aspects. However, the completeness of the classification is very limited. For example, TEclass can only classify TEs into the following four classes: DNA transposons, LTR, LINE, and SINE, and its classification results tend to have high false-positive rates. Moreover, DeepTE uses CNNs to classify unknown TEs by converting sequences into input vectors based on k-mer counting, which can be used to distinguish TEs and non-TEs with relatively low false-positive rates. Both TEclass and REPCLASS cannot distinguish between TEs and other non-TEs, so DeepTE is superior to them. Nevertheless, DeepTE is also not perfect. First, the completeness of its classification remains unsatisfactory. Second, DeepTE is not specifically designed to classify nested TE, and the databases it depends on do not include annotations for nested TEs. Deep neural networks (DNNs) have great application potential in automated repeat classification. However, current methods did not maximize the advantages of DNNs. Therefore, developing superior DNNs and models for more comprehensive and accurate repeat classification is one of the main research focuses for the future.
TEs carry cis-regulatory sequences that can alter gene regulatory networks through redistributing transcription factor binding sites and developing novel enhancer activities. Its abnormal expression is closely related to many complex diseases, such as cancers. However, the role of TEs in cell-type heterogeneity and biological processes has not been fully revealed, and research in this field is still in its infancy. With the rapid development of single-cell technologies, scRNA-seq has become an efficient method for observing cell activity, which can be used to analyze gene-centric and TE expression accurately. Therefore, a future research focus is to quantify TE expression and explore the role of TEs in the pathway and mechanism of complex diseases at the single-cell level.
Conclusion
Repetitive DNA sequences play an indispensable role in the physiological activities of organisms, and they comprise almost half of the human genome. Repeats in genomes can be divided into TEs and TRs. TEs can result in mutations, altered gene expression, chromosome rearrangement. etc., which are related to many diseases, such as cancers, genetic disorders, autoimmune diseases, and metabolic disorders. TRs, especially STRs, are highly variable, which can accelerate the gene expression evolution and generate sufficient variability that allows a rapid evolution and adaptation of organisms, and play a vital role in the structural stability of genetic materials and regulate gene expression, causing various disorders. Due to a lack of sufficiently advanced detection technologies, the role and effect of repeats in genomes, especially the human genome, have been underestimated. We believe that this review will be helpful in the understanding of repeats in genomes and provide guidance for repeat annotation (detection, classification, and masking) and in-depth exploration of its association with human diseases.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The reference genomes of six species: Homo sapiens (GCF_000001405.39), Gallus (GCF_016699485.2), Mouse (GCF_000001635.27), Drosophila melanogaster (GCA_018903765.1), Glycine max (GCA_000004515.5) and Leafcutter ant (GCA_000204515.1) are downloaded from the NCBI website (https://www.ncbi.nlm.nih.gov/). Five groups of NGS short reads: Leafcutter Ant (ERR034186, https://www.ncbi.nlm.nih.gov/), D.melanogaster (SRR350 908, https://www.ncbi.nlm.nih.gov/), Mouse (ERR2894257, https://www.ncbi.nlm.nih.gov/), Human-chr14(https://gage.cbcb.umd.edu/) and HG003_24149_father (D2 S2 L001 R1 001, ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data), three groups of barcode linked reads (HG003_24149_father, HG004_NA24143, and HG002_NA24385_son, ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data), three groups of CCS long reads (HG003_24149_father, HG004_NA24143_mother and HG002_NA24385_son, ftp://ftp-trace.ncbi.nlm.nih.gov/giab/ftp/data), and four groups of PacBio long reads (dro_100k, human_100k, dmel_filtered and human_polished, https://github.com/ruiguo-bio/replong) are used to evaluate the performance of each tool in this study.
References
Biscotti, M. A., Olmo, E. & Heslop-Harrison, J. S. Repetitive DNA in eukaryotic genomes. Chromosom. Res. 23, 415–420 (2015).
Mrázek, J., Guo, X. & Shah, A. Simple sequence repeats in prokaryotic genomes. Proc. Natl Acad. Sci. USA. 104, 8472–8477 (2007).
Jurka, J., Kapitonov, V. V., Kohany, O. & Jurka, M. V. Repetitive sequences in complex genomes: structure and evolution. Annu. Rev. Genom. Hum. Genet. 8, 241–259 (2007).
Treangen, T. J., Abraham, A. L., Touchon, M. & Rocha, E. P. Genesis, effects and fates of repeats in prokaryotic genomes. FEMS Microbiol. Rev. 33, 539–571 (2009).
Bernabe, I. B. et al. Genome-wide contribution of common short-tandem repeats to Parkinson’s disease genetic risk. Brain 146, 65–74 (2023).
Nekrutenko, A. & Li, W. H. Transposable elements are found in a large number of human protein-coding genes. Trends Genet. 17, 619–621 (2001).
Alexander, R. P., Fang, G., Rozowsky, J., Snyder, M. & Gerstein, M. B. Annotating non-coding regions of the genome. Nat. Rev. Genet. 11, 559–571 (2010).
Bourque, G. et al. Ten things you should know about transposable elements. Genome Biol. 19, 199 (2018).
Zhang, X. & Meyerson, M. Illuminating the noncoding genome in cancer. Nat. Cancer 1, 864–872 (2020).
Mehrotra, S. & Goyal, V. Repetitive Sequences in Plant Nuclear DNA: Types, Distribution, Evolution and Function. Genom. Proteom. Bioinform. 12, 164–171 (2014).
Zu, T. et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl Acad. Sci. USA. 108, 260–5 (2011).
Al-Turki, T. M. & Griffith, J. D. Mammalian telomeric RNA (TERRA) can be translated to produce valine-arginine and glycine-leucine dipeptide repeat proteins. Proc. Natl Acad. Sci. USA. 120, e2221529120 (2023).
Hannan, A. J. Tandem repeats mediating genetic plasticity in health and disease. Nat. Rev. Genet. 19, 286–298 (2018).
Ishiura, H. et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 51, 1222–1232 (2019).
Shah, N. M. et al. Pan-cancer analysis identifies tumor-specific antigens derived from transposable elements. Nat. Genet. 55, 631–639 (2023). This article reported that cryptic promoters within transposable elements (TEs) can be transcriptionally reactivated in tumors to create new TE-chimeric transcripts, which can produce immunogenic antigens.
Touati, R. et al. New methodology for repetitive sequences identification in human X and Y chromosomes. Biomed. Signal Proc. Control 64, 102207 (2021).
Novák, P. et al. TAREAN: a computational tool for identification and characterization of satellite DNA from unassembled short reads. Nucleic Acids Res. 45, e111–e111 (2017).
Liehr, T. Repetitive elements in humans. Int. J. Mol. Sci. 22, 2072 (2021).
Novák, P., Neumann, P. & Macas, J. Global analysis of repetitive DNA from unassembled sequence reads using RepeatExplorer2. Nat. Protoc. 15, 3745–3776 (2020).
McNulty, S. M. & Sullivan, B. A. Alpha satellite DNA biology: finding function in the recesses of the genome. Chromosom. Res. 26, 115–138 (2018).
Youssef, N., Budd, A. & Bielawski, J. P. Introduction to Genome Biology and Diversity. Methods Mol. Biol. 1910, 3–31 (2019).
Bishop, C. E., Guellaen, G., Geldwerth, D. VossR., Fellous, M. & Weissenbach, J. Single-copy DNA sequences specific for the human Y chromosome. Nature 303, 831–832 (1983).
Hou, Z., Romero, R., Uddin, M., Than, N. G. & Wildman, D. E. Adaptive history of single copy genes highly expressed in the term human placenta. Genomics 93, 33–41 (2009).
Pavlicek A., Kapitonov V.V., & Jurka J. Human Repetitive DNA[M]. Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine. (Springer, Berlin, Heidelberg, 2005).
Kojima, K. K. Structural and sequence diversity of eukaryotic transposable elements. Genes Genet. Syst. 94, 233–252 (2020).
Genovese, L. M. et al. A Census of Tandemly Repeated Polymorphic Loci in Genic Regions Through the Comparative Integration of Human Genome Assemblies. Front. Genet. 9, 155 (2018).
Richard, G. F., Kerrest, A. & Dujon, B. Comparative genomics and molecular dynamics of DNA repeats in eukaryotes. Microbiol. Mol. Biol. Rev. 72, 686–727 (2008).
Sullivan, L. L., Chew, K. & Sullivan, B. A. α satellite DNA variation and function of the human centromere. Nucleus 8, 331–339 (2017).
Sawaya, S. et al. Microsatellite tandem repeats are abundant in human promoters and are associated with regulatory elements. PLoS ONE 8, e54710 (2013).
Richard, G. F. & Pâques, F. Mini- and microsatellite expansions: the recombination connection. EMBO Rep. 1, 122–126 (2000).
Li, H. Identifying centromeric satellites with dna-brnn. Bioinformatics 35, 4408–4410 (2019).
Alaguponniah, S. et al. Finding of novel telomeric repeats and their distribution in the human genome. Genomics 112, 3565–3570 (2020).
Riethman, H. Human subtelomeric copy number variations. Cytogenet. Genome Res. 123, 244–252 (2008).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199 (2010).
Kojima, K. K. & Jurka, J. Crypton transposons: identification of new diverse families and ancient domestication events. Mobile DNA 2, 12 (2011).
Krupovic, M. & Koonin, E. V. Polintons: a hotbed of eukaryotic virus, transposon and plasmid evolution. Nat. Rev. Microbiol. 13, 105–115 (2015).
Lee, T. F. et al. RNA polymerase V-dependent small RNAs in Arabidopsis originate from small, intergenic loci including most SINE repeats. Epigenetics 7, 781–795 (2012).
Pace, J. K. & Feschotte, C. The evolutionary history of human DNA transposons: evidence for intense activity in the primate lineage. Genome Res. 17, 422–432 (2007).
Muñoz-López, M. & Garcĺa-Pérez, J. L. DNA transposons: nature and applications in genomics. Curr. Genom. 11, 115–128 (2010).
Kojima, K. K. Human transposable elements in Repbase: genomic footprints from fish to humans. Mobile DNA 9, 2 (2018).
David, J. F. Retrotransposons. Curr. Biol. 22, R432–R437 (2012).
Muszewska, A., Hoffman-Sommer, M. & Grynberg, M. LTR retrotransposons in fungi. PLoS ONE 6, e29425 (2011).
Thompson, P. J., Macfarlan, T. S. & Lorincz, M. C. Long Terminal Repeats: From Parasitic Elements to Building Blocks of the Transcriptional Regulatory Repertoire. Mol. Cell 62, 766–76 (2016).
Ardeljan, D., Taylor, M. S., Ting, D. T. & Burns, K. H. The Human Long Interspersed Element-1 Retrotransposon: An Emerging Biomarker of Neoplasia. Clin. Chem. 63, 816–822 (2017).
Kramerov, D. A. & Vassetzky, N. S. Origin and evolution of SINEs in eukaryotic genomes. Heredity 107, 487–495 (2011).
Han, G. et al. Diversity of short interspersed nuclear elements (SINEs) in lepidopteran insects and evidence of horizontal SINE transfer between baculovirus and lepidopteran hosts. BMC Genom. 22, 226 (2021).
Malicki, M., Spaller, T., Winckler, T. & Hammann, C. DIRS retrotransposons amplify via linear, single-stranded cDNA intermediates. Nucleic Acids Res. 48, 4230–4243 (2020).
Wiegand, S. et al. The Dictyostelium discoideum RNA-dependent RNA polymerase RrpC silences the centromeric retrotransposon DIRS-1 post-transcriptionally and is required for the spreading of RNA silencing signals. Nucleic Acids Res. 42, 3330–3345 (2014).
Wang, Y., Gallagher-Jones, M., Suśac, L., Song, H. & Feigon, J. A structurally conserved human and Tetrahymena telomerase catalytic core. Proc. Natl Acad. Sci. USA. 117, 31078–31087 (2020).
Arkhipova, I. R. Distribution and Phylogeny of Penelope-Like Elements in Eukaryotes. Syst. Biol. 55, 875–885 (2006).
Gladyshev, E. A. & Arkhipova, I. R. Telomere-associated endonuclease-deficient Penelope-like retroelements in diverse eukaryotes. Proc. Natl Acad. Sci. USA. 104, 9352–9357 (2007).
Han, J. S. Non-long terminal repeat (non-LTR) retrotransposons: mechanisms, recent developments, and unanswered questions. Mobile DNA 1, 15 (2010).
Scott, E. C. et al. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Res. 26, 745–755 (2016).
Miki, Y. et al. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 52, 643–645 (1992).
Larsen, P. A. et al. The Alu neurodegeneration hypothesis: A primate-specific mechanism for neuronal transcription noise, mitochondrial dysfunction, and manifestation of neurodegenerative disease. Alzheimers Dement. 13, 828–838 (2017).
Payer, L. M. et al. Structural variants caused by Alu insertions are associated with risks for many human diseases. Proc. Natl Acad. Sci. USA. 114, E3984–E3992 (2017).
Gianfrancesco, O., Bubb, V. J. & Quinn, J. P. SVA retrotransposons as potential modulators of neuropeptide gene expression. Neuropeptides 64, 3–7 (2017).
Petrozziello, T. et al. SVA insertion in X-linked Dystonia Parkinsonism alters histone H3 acetylation associated with TAF1 gene. PLoS ONE 15, e0243655 (2020).
Lerat, E. & Capy, P. Retrotransposons and retroviruses: analysis of the envelope gene. Mol. Biol. Evol. 16, 1198–1207 (1999).
Havecker, E. R., Gao, X. & Voytas, D. F. The diversity of LTR retrotransposons. Genome Biol. 5, 225 (2004).
Gro˙ger, V. et al. Formation of HERV-K and HERV-Fc1 Envelope Family Members is Suppressed on Transcriptional and Translational Level. Int. J. Mol. Sci. 21, 7855 (2020).
Nelson, P. N. et al. Human endogenous retroviruses: transposable elements with potential? Clin. Exp. Immunol. 138, 1–9 (2004).
Zhao, J. et al. Expression of Human Endogenous Retrovirus Type K Envelope Protein is a Novel Candidate Prognostic Marker for Human Breast Cancer. Genes Cancer 2, 914–922 (2011).
Sohn, J. & Nam, J. W. The present and future of de novo whole-genome assembly. Brief Bioinform. 19, 23–40 (2018).
Liao, X. et al. Current challenges and solutions of de novo assembly. Quant. Biol. 7, 90–109 (2019).
Kamath, G. M. et al. HINGE: long-read assembly achieves optimal repeat resolution. Genome Res. 27, 747–756 (2017). This article reported an assembler that seeks to achieve optimal repeat resolution by distinguishing repeats that can be resolved given the data from those that cannot.
Jain, C. et al. Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods 19, 705–710 (2022).
Jakubosky, D. et al. Properties of structural variants and short tandem repeats associated with gene expression and complex traits. Nat. Commun. 11, 2927 (2020).
Liao, X. et al. Improving de novo assembly based on read classification. IEEE/ACM Trans. Comput. Biol. Bioinform. 17, 177–188 (2018).
Miga, K. H. et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature 585, 79–84 (2020).
Narzisi, G. & Schatz, M. C. The challenge of small-scale repeats for indel discovery. Front. Bioeng. Biotechnol. 3, 8 (2015).
Trigiante, G., Blanes, R. N. & Cerase, A. Emerging Roles of Repetitive and Repeat-Containing RNA in Nuclear and Chromatin Organization and Gene Expression. Front. Cell Dev. Biol. 9, 735527 (2021).
Gao, D. et al. Transposons play an important role in the evolution and diversification of centromeres among closely related species. Front. Plant Sci. 6, 216 (2015).
Nishihara, H. Transposable elements as genetic accelerators of evolution: contribution to genome size, gene regulatory network rewiring and morphological innovation. Genes Genet. Syst. 94, 269–281 (2020).
Ramakrishnan, M. et al. The Dynamism of Transposon Methylation for Plant Development and Stress Adaptation. Int. J. Mol. Sci. 22, 11387 (2021).
Chuong, E. B., Elde, N. C. & Feschotte, C. Regulatory activities of transposable elements: from conflicts to benefits. Nat. Rev. Genet. 18, 71–86 (2017).
González, J. et al. High rate of recent transposable element-induced adaptation in Drosophila melanogaster. PLoS Biol. 6, e251 (2008).
Ayarpadikannan, S. & Kim, H. S. The impact of transposable elements in genome evolution and genetic instability and their implications in various diseases. Genom. Inform. 12, 98–104 (2014).
Hancks, D. C. & Kazazian, H. H. Roles for retrotransposon insertions in human disease. Mobile DNA 7, 9 (2016).
Voronova, A. et al. Retrotransposon distribution and copy number variation in gymnosperm genomes. Tree Genet. Genomes 13, 88 (2017).
Pavlicek, A., Gentles, A. J., Paces, J., Paces, V. & Jurka, J. Retroposition of processed pseudogenes: the impact of RNA stability and translational control. Trends Genet. 22, 69–73 (2006).
Ovchinnikov, I., Troxel, A. B. & Swergold, G. D. Genomic characterization of recent human LINE-1 insertions: evidence supporting random insertion. Genome Res. 11, 2050–2058 (2001).
Ponomaryova, A. A. et al. Aberrant Methylation of LINE-1 Transposable Elements: A Search for Cancer Biomarkers. Cells 9, 2017 (2020).
McKerrow, W. et al. LINE-1 expression in cancer correlates with p53 mutation, copy number alteration, and S phase checkpoint. Proc. Natl Acad. Sci. USA. 119, e2115999119 (2022). This article reported that LINE-1 expression in cancer correlates with p53 mutation, copy number alteration, and S phase checkpoint.
Witherspoon, D. J. et al. Mobile element scanning (ME-Scan) identifies thousands of novel Alu insertions in diverse human populations. Genome Res. 23, 107–116 (2013).
Savage, A. L. et al. Characterisation of retrotransposon insertion polymorphisms in whole genome sequencing data from individuals with amyotrophic lateral sclerosis. Gene 843, 146799 (2022).
Zhang, Y. et al. Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat. Genet. 51, 1380–1388 (2019).
Uzunović, J., Josephs, E. B., Stinchcombe, J. R. & Wright, S. I. Transposable Elements Are Important Contributors to Standing Variation in Gene Expression in Capsella Grandiflora. Mol. Biol. Evol. 36, 1734–1745 (2019).
Chishima, T., Iwakiri, J. & Hamada, M. Identification of Transposable Elements Contributing to Tissue-Specific Expression of Long Non-Coding RNAs. Genes 9, 23 (2018).
Horváth, V., Merenciano, M. & González, J. Revisiting the Relationship between Transposable Elements and the Eukaryotic Stress Response. Trends Genet. 33, 832–841 (2017).
Anastasia, A. Z. et al. Transcriptional regulation of human-specific SVAF1 retrotransposons by cis-regulatory MAST2 sequences. Gene 505, 128–136 (2012).
Barnada, S. M. et al. Genomic features underlie the co-option of SVA transposons as cis-regulatory elements in human pluripotent stem cells. PLoS Genet. 18, e1010225 (2022).
Zhang, X. O., Gingeras, T. R. & Weng, Z. Genome-wide analysis of polymerase III-transcribed Alu elements suggests cell-type-specific enhancer function. Genome Res. 29, 1402–1414 (2019).
Lupski, J. R. & Stankiewicz, P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 1, e49 (2005).
Cordaux, R. & Batzer, M. A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 10, 691–703 (2009).
Klein, S. J. & O’Neill, R. J. Transposable elements: genome innovation, chromosome diversity, and centromere conflict. Chromosom. Res. 26, 5–23 (2018).
Burns, K. Transposable elements in cancer. Nat. Rev. Cancer 17, 415–424 (2017). This article reported that the activity of transposable elements in human cancers, particularly long interspersed element-1 (LINE-1), leads to somatically acquired insertions in cancer genomes.
Ahmadi, A. et al. Transposable elements in brain health and disease. Ageing Res. Rev. 64, 101153 (2020). This article reported that TEs are expressed and active in the brain, challenging the dogma that neuronal genomes are static and revealing that they are susceptible to somatic genomic alterations, and have a role in behavior and cognition.
Saleh, A., Macia, A. & Muotri, A. R. Transposable Elements, Inflammation, and Neurological Disease. Front. Neurol. 10, 894 (2019).
Kim, Y. J., Lee, J. & Han, K. Transposable Elements: No More ’Junk DNA’. Genom. Inform. 10, 226–233 (2012).
Balachandran, P. et al. Transposable element-mediated rearrangements are prevalent in human genomes. Nat. Commun. 13, 7115 (2022). This article reported that the transposable element-mediated rearrangements are enriched in genic loci and can create potentially important risk alleles such as a deletion in TRIM65, a known cancer biomarker and therapeutic target.
Niu, Y. et al. Characterizing mobile element insertions in 5675 genomes. Nucleic Acids Res. 50, 2493–2508 (2022).
Huang, C. R., Burns, K. H. & Boeke, J. D. Active transposition in genomes. Annu. Rev. Genet. 46, 651–675 (2012).
Cordaux, R., Hedges, D. J., Herke, S. W. & Batzer, M. A. Estimating the retrotransposition rate of human Alu elements. Gene 373, 134–137 (2006).
Rosser, J. M. & An, W. L1 expression and regulation in humans and rodents. Front. Biosci. (Elite Ed) 4, 2203–2225 (2012).
Chuang, N. T. et al. Mutagenesis of human genomes by endogenous mobile elements on a population scale. Genome Res. 31, 2225–35 (2021).
Payer, L. M. & Burns, K. H. Transposable elements in human genetic disease. Nat. Rev. Genet. 20, 760–772 (2019). This article reviewed many ways human retrotransposons contribute to genome function, their dysregulation in diseases including cancer, and how they affect genetic disease.
Kannan, S. et al. Transposable Element Insertions in Long Intergenic Non-Coding RNA Genes. Front. Bioeng. Biotechnol. 3, 71 (2015).
Etchegaray, E., Naville, M., Volff, J. N. & Haftek-Terreau, Z. Transposable element-derived sequences in vertebrate development. Mob. DNA 12, 1 (2021).
Johnson, R. & Guigó, R. The RIDL hypothesis: transposable elements as functional domains of long noncoding RNAs. RNA 20, 959–976 (2014).
Cuevas-Diaz, D. R. et al. Long non-coding RNAs: important regulators in the development, function and disorders of the central nervous system. Neuropathol. Appl. Neurobiol. 45, 538–556 (2019).
Grandi, N. & Tramontano, E. HERV Envelope Proteins: Physiological Role and Pathogenic Potential in Cancer and Autoimmunity. Front. Microbiol. 9, 462 (2018).
Mao, J., Zhang, Q. & Cong, Y. S. Human endogenous retroviruses in development and disease. Comput. Struct. Biotechnol. J. 19, 5978–5986 (2021).
Hermant, C. & Torres-Padilla, M. E. TFs for TEs: the transcription factor repertoire of mammalian transposable elements. Genes Dev. 35, 22–39 (2021).
Senft, A. D. & Macfarlan, T. S. Transposable elements shape the evolution of mammalian development. Nat. Rev. Genet. 22, 691–711 (2021).
Evrony, G. D. et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496 (2012).
Ali, A., Han, K. & Liang, P. Role of transposable elements in gene regulation in the human genome. Life 11, 118 (2021).
Fueyo, R., Judd, J., Feschotte, C. & Wysocka, J. Roles of transposable elements in the regulation of mammalian transcription. Nat. Rev. Mol. Cell Biol. 23, 481–497 (2022). This article reported that TEs often contain sequences capable of recruiting the host transcription machinery, which they use to express their own products and promote transposition.
Usdin, K. The biological effects of simple tandem repeats: lessons from the repeat expansion diseases. Genome Res. 18, 1011–1019 (2008).
Haubold, B. & Wiehe, T. How repetitive are genomes? BMC Bioinform. 7, 541–551 (2006).
Yi, H. et al. The Tandem Repeats Enabling Reversible Switching between the Two Phases of β-Lactamase Substrate Spectrum. PLOS Genet. 10, e1004640 (2014).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
O’Dushlaine, C. T., Edwards, R. J., Park, S. D. & Shields, D. C. Tandem repeat copy-number variation in protein-coding regions of human genes. Genome Biol. 6, R69 (2005).
Hannan, A. J. Tandem repeat polymorphisms: Mediators of genetic plasticity modulators of biological diversity and dynamic sources of disease susceptibility. Adv. Exp. Med. Biol. 769, 1–9 (2012).
Fan, H. & Chu, J. Y. A brief review of short tandem repeat mutation. Genom. Proteom. Bioinform. 5, 7–14 (2007).
Castillo-Lizardo, M., Henneke, G. & Viguera, E. Replication slippage of the thermophilic DNA polymerases B and D from the Euryarchaeota Pyrococcus abyssi. Front. Microbiol. 5, 403 (2014).
Gymrek, M., Willems, T., Reich, D. & Erlich, Y. Interpreting short tandem repeat variations in humans using mutational constraint. Nat. Genet. 49, 1495–1501 (2017).
Gemayel, R., Vinces, M. D., Legendre, M. & Verstrepen, K. J. Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annu. Rev. Genet. 44, 445–477 (2010).
Mukamel, R. E. et al. Protein-coding repeat polymorphisms strongly shape diverse human phenotypes. Science 373, 1499–1505 (2021).
Farré, M., Bosch, M., López-Giráldez, F., Ponsá, M. & Ruiz-Herrera, A. Assessing the role of tandem repeats in shaping the genomic architecture of great apes. PLoS ONE 6, e27239 (2011).
Gemayel, R., Cho, J., Boeynaems, S. & Verstrepen, K. J. Beyond junk-variable tandem repeats as facilitators of rapid evolution of regulatory and coding sequences. Genes 3, 461–80 (2012).
Shi, Y. et al. Characterization of genome-wide STR variation in 6487 human genomes. Nat. Commun. 14, 2092 (2023). This article reported that short tandem repeat mutations were affected by motif length, chromosome context and epigenetic features.
Fotsing, S. F. et al. The impact of short tandem repeat variation on gene expression. Nat. Genet. 51, 1652–1659 (2019). This article reported that expression of short tandem repeats explain a sizable portion (10–15%) of the cis heritability of gene expression.
Aguilar, M. & Prieto, P. Telomeres and Subtelomeres Dynamics in the Context of Early Chromosome Interactions During Meiosis and Their Implications in Plant Breeding. Front. Plant Sci. 12, 672489 (2021).
Lamb, J. C. & Birchler, J. A. The role of DNA sequence in centromere formation. Genome Biol. 4, 214 (2003).
Miga, K. H. & Alexandrov, I. A. Variation and evolution of human centromeres: a field guide and perspective. Ann. Rev. Genet. 55, 583–602 (2021).
Lim, C. J. & Cech, T. R. Shaping human telomeres: from shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 22, 283–298 (2021).
Sun, J. H. et al. Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 175, 224-238.e15 (2018).
Ishiura, H. et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 50, 581–590 (2018).
Albertin, C. B. et al. The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 524, 220–224 (2015).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9orf72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Duan, Y. et al. PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 29, 233–247 (2019).
Raghupathy, N. & Durand, D. Gene cluster statistics with gene families. Mol. Biol. Evol. 26, 957–968 (2009).
Bonthala, V. S. & Stich, B. Genetic Divergence of Lineage-Specific Tandemly Duplicated Gene Clusters in Four Diploid Potato Genotypes. Front. Plant Sci. 13, 875202 (2022).
Kuzmin, E., Taylor, J. S. & Boone, C. Retention of duplicated genes in evolution. Trends Genet. 38, 59–72 (2022).
Sultanov, D. & Hochwagen, A. Varying strength of selection contributes to the intragenomic diversity of rRNA genes. Nat. Commun. 13, 7245 (2022).
Blokhina, Y. P. & Buchwalter, A. Moving fast and breaking things: Incidence and repair of DNA damage within ribosomal DNA repeats. Mutat. Res. 821, 111715 (2020).
Pajic, P. et al. A mechanism of gene evolution generating mucin function. Sci. Adv. 8, eabm8757 (2022).
Gymrek, M. et al. Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet. 48, 22–29 (2016).
Malik, I., Kelley, C. P., Wang, E. T. & Todd, P. K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 22, 589–607 (2021).
Trost, B. et al. Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature 586, 80–86 (2020).
Chintalaphani, S. R. et al. An update on the neurological short tandem repeat expansion disorders and the emergence of long-read sequencing diagnostics. Acta Neuropathol. Commun. 9, 98 (2021).
Depienne, C. & Mandel, J. L. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 108, 764–785 (2021). This article reported the development and remaining challenges of the field of repeat expansion disorders over the past 30 years.
Goodman, L. D. & Bonini, N. M. New Roles for Canonical Transcription Factors in Repeat Expansion Diseases. Trends Genet. 36, 81–92 (2020).
Chen, W., Swanson, B. J. & Frankel, W. L. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn. Pathol. 12, 24 (2017).
Taylor, J. P., Brown Jr, R. H. & Cleveland, D. W. Decoding ALS: from genes to mechanism. Nature 539, 197–206 (2016).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 11–17 (2015).
Hubley, R. et al. The Dfam database of repetitive DNA families. Nucleic Acids Res. 44, D81–D89 (2016).
Liao, X. et al. msRepDB: a comprehensive repetitive sequence database of over 80 000 species. Nucleic Acids Res. 50, D236–D245 (2021).
Neumann, P. et al. Systematic survey of plant LTR-retrotransposons elucidates phylogenetic relationships of their polyprotein domains and provides a reference for element classification. Mobile DNA 10, 1–18 (2019).
Jaina, M. et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419 (2021).
Scott, M. & Thomas, L. M. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 32, W20–W25 (2004).
Jurka, J., Klonowski, P., Dagman, V. & Pelton, P. CENSOR-a program for identification and elimination of repetitive elements from DNA sequences. Comput. Chem. 20, 119–121 (1996).
Kennedy, R. C. et al. An automated homology-based approach for identifying transposable elements. BMC Bioinform. 12, 130 (2011).
Li, X., Kahveci, T. & Settles, A. M. A novel genome-scale repeat finder geared towards transposons. Bioinformatics 24, 468–476 (2007).
Fiston-Lavier, A. S., Carrigan, M., Petrov, D. A. & González, J. T-lex: a program for fast and accurate assessment of transposable element presence using next-generation sequencing data. Nucleic Acids Res. 39, e36 (2010).
Wicker, T. et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 8, 973–982 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 9, 18 (2008). This article reported LTRharvest, currently the most well-known LTR retrotransposon detection tool.
Darzentas, N., Bousios, A., Apostolidou, V. & Tsaftaris, A. S. MASiVE: Mapping and Analysis of SireVirus Elements in plant genome sequences. Bioinformatics 26, 2452–2454 (2010).
Rho, M., Choi, J. H., Kim, S., Lynch, M. & Tang, H. De novo identification of LTR retrotransposons in eukaryotic genomes. BMC Genom. 8, 90 (2007).
Matej, L., Pavel, J., Ivan, V., Michal, C. & Eduard, K. TE-greedy-nester: structure-based detection of LTR retrotransposons and their nesting. Bioinformatics 36, 4991–4999 (2020).
Wenke, T. et al. Targeted identification of short interspersed nuclear element families shows their widespread existence and extreme heterogeneity in plant genomes. Plant Cell 23, 3117–3128 (2011).
Hongliang, M. & Hao, W. SINE_scan: an efficient tool to discover short interspersed nuclear elements (SINEs) in large-scale genomic datasets. Bioinformatics 33, 743–745 (2017).
Li, Y., Jiang, N. & Sun, Y. AnnoSINE: a short interspersed nuclear elements annotation tool for plant genomes. Plant Physiol. 188, 955–970 (2022).
Tu, Z. Eight novel families of miniature inverted repeat transposable elements in the African malaria mosquito Anopheles gambiae. Proc. Natl. Acad. Sci. USA. 98, 1699–1704 (2001).
Chen, Y., Zhou, F., Li, G. & Xu, Y. MUST: a system for identification of miniature inverted-repeat transposable elements and applications to Anabaena variabilis and Haloquadratum walsbyi. Gene 436, 1–7 (2009).
Ye, C., Ji, G. & Liang, C. detectMITE: A novel approach to detect miniature inverted repeat transposable elements in genomes. Sci. Rep. 6, 19688 (2016).
Yang, G. MITE Digger, an efficient and accurate algorithm for genome wide discovery of miniature inverted repeat transposable elements. BMC Bioinform. 14, 186 (2013).
Crescente, J. M., Zavallo, D., Helguera, M. & Vanzetti, L. S. MITE Tracker: an accurate approach to identify miniature inverted-repeat transposable elements in large genomes. BMC Bioinform. 19, 348 (2018).
Lerat, E. Identifying repeats and transposable elements in sequenced genomes: how to find your way through the dense forest of programs. Heredity 104, 520–533 (2010).
Agarwal, P. & States, D. J. The Repeat Pattern Toolkit (RPT): analyzing the structure and evolution of the C. elegans genome. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2, 1–9 (1994).
Chen, G. L., Chang, Y. J. & Hsueh, C. H. PRAP: an ab initio software package for automated genome-wide analysis of DNA repeats for prokaryotes. Bioinformatics 29, 2683–2689 (2013).
Robert, C. E. & Eugene, W. M. PILER: identification and classification of genomic repeats. Bioinformatics 21, i152–i158 (2005).
Nicolas, J., Tempel, S., Fiston-Lavier, A. S. & Cherif, E. Finding and characterizing repeats in plant genomes. Methods Mol. Biol. 2443, 327–385 (2016).
Liao, X. et al. A sensitive repeat identification framework based on short and long reads. Nucleic Acids Res. 49, e100–e100 (2021).
Saha, S., Bridges, S., Magbanua, Z. V. & Peterson, D. G. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 36, 2284–2294 (2008).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Li, R. et al. ReAS: Recovery of ancestral sequences for transposable elements from the unassembled reads of a whole genome shotgun. PLoS Comput. Biol. 1, e43 (2005).
Shi, J. & Liang, C. Generic Repeat Finder: A High-Sensitivity Tool for Genome-Wide De Novo Repeat Detection. Plant. Physiol. 180, 1803–1815 (2019).
Koch, P., Platzer, M. & Downie, B. R. RepARK-de novo creation of repeat libraries from whole-genome NGS reads. Nucleic Acids Res. 42, e80–e80 (2014).
Chu, C., Nielsen, R. & Wu, Y. REPdenovo: inferring de novo repeat motifs from short sequence reads. PloS ONE 11, e0150719 (2016).
Liao, X., Gao, X., Zhang, X., Wu, F. X. & Wang, J. RepAHR: an improved approach for de novo repeat identification by assembly of the high-frequency reads. BMC Bioinform. 21, 463 (2020).
Guo, R. et al. RepLong: de novo repeat identification using long read sequencing data. Bioinformatics 34, 1099–1107 (2017).
Kolpakov, R., Bana, G. & Kucherov, G. mreps: Efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res. 31, 3672–8 (2003).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–80 (1999). This article reported tandem repeat finder (TRF), currently the most well-known tandem repeat detection tool.
Jorda, J. & Kajava, A. V. T-REKS: identification of Tandem REpeats in sequences with a K-meanS based algorithm. Bioinformatics 25, 2632–8 (2009).
Wlodzimierz, P., Hong, M. & Henderson, I. R. TRASH: Tandem Repeat Annotation and Structural Hierarchy. Bioinformatics 39, btad308 (2023).
Jam H. Z. et al. A deep population reference panel of tandem repeat variation. bioRxiv 2023.03.09.531600, 1–37 (2023).
Fazal S. et al. RExPRT: a machine learning tool to predict pathogenicity of tandem repeat loci. bioRxiv 2023.03.22.533484, 1–30 (2023).
Dolzhenko, E. et al. ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35, 4754–4756 (2019).
Mousavi, N., Shleizer-Burko, S., Yanicky, R. & Gymrek, M. Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acids Res. 47, e90 (2019).
Dolzhenko, E. et al. ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol. 21, 1–14 (2020).
Chiu, R. et al. Straglr: discovering and genotyping tandem repeat expansions using whole genome long-read sequences. Genome Biol. 22, 224 (2021).
Dashnow, H. et al. STRling: a k-mer counting approach that detects short tandem repeat expansions at known and novel loci. Genome Biol. 23, 257 (2022).
Ou, S. et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline. Genome Biol. 20, 275 (2019).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA. 117, 9451–9457 (2020).
Budiš, J. et al. Dante: genotyping of known complex and expanded short tandem repeats. Bioinformatics 35, 1310–1317 (2019).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass-a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330 (2009).
Hoede, C. et al. PASTEC: an automatic transposable element classification tool. PLoS ONE 9, e91929 (2014).
Feschotte, C. et al. Exploring repetitive DNA landscapes using REPCLASS, a tool that automates the classification of transposable elements in eukaryotic genomes. Genome Biol. Evol. 1, 205–220 (2009).
Mor, B., Garhwal, S. & Kumar, A. A Systematic Review of Hidden Markov Models and Their Applications. Arch. Computat. Methods Eng. 28, 1429–1448 (2021).
Yan, H., Bombarely, A. & Li, S. DeepTE: a computational method for de novo classification of transposons with convolutional neural network. Bioinformatics 36, 4269–4275 (2020).
da Cruz, M. H. P. et al. TERL: classification of transposable elements by convolutional neural networks. Brief Bioinform. 22, bbaa185 (2021).
Martinez-Gomez, L. et al. Few SINEs of life: Alu elements have little evidence for biological relevance despite elevated translation. NAR Genom. Bioinform. 2, lqz023 (2020).
Salem, A. H. et al. Recently integrated Alu elements and human genomic diversity. Mol. Biol. Evol. 20, 1349–1361 (2003).
Hancks, D. C. & Kazazian Jr, H. H. SVA retrotransposons: Evolution and genetic instability. Semin Cancer Biol. 20, 234–245 (2010).
Hancks, D. C. et al. The minimal active human SVA retrotransposon requires only the 5’-hexamer and Alu-like domains. Mol. Cell Biol. 32, 4718–4726 (2012).
Beck, C. R. et al. LINE-1 retrotransposition activity in human genomes. Cell 141, 1159–1170 (2010).
Grandi, N. & Tramontano, E. Human Endogenous Retroviruses Are Ancient Acquired Elements Still Shaping Innate Immune Responses. Front. Immunol. 9, 2039 (2018).
Buzdin, A. et al. Human-specific subfamilies of HERV-K (HML-2) long terminal repeats: three master genes were active simultaneously during branching of hominoid lineages. Genomics 81, 149–156 (2003).
van Bree, E. J. et al. A hidden layer of structural variation in transposable elements reveals potential genetic modifiers in human disease-risk loci. Genome Res. 32, 656–670 (2022).
Poggi, L. et al. Differential efficacies of Cas nucleases on microsatellites involved in human disorders and associated off-target mutations. Nucleic Acids Res. 49, 8120–8134 (2021).
Annear, D. J. et al. Non-Mendelian inheritance patterns and extreme deviation rates of CGG repeats in autism. Genome Res. 32, 1967–1980 (2022).
Irigoyen, A. M. et al. Differential expression of the androgen receptor gene is correlated with CAG polymorphic repeats in patients with prostate cancer. J. Genet. 102, 23 (2023).
Mu˙ller, N. A. et al. A single gene underlies the dynamic evolution of poplar sex determination. Nat. Plants 6, 630–637 (2020).
Kapitonov, V. V. & Jurka, J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet. 9, 411–412 (2008).
Albert, P. S. et al. Whole-chromosome paints in maize reveal rearrangements, nuclear domains, and chromosomal relationships. Proc. Natl. Acad. Sci. USA. 116, 1679–1685 (2019).
Qian, Z. et al. The chromosome level genome of a free floating aquatic weed Pistia stratiotes provides insights into its rapid invasion. Mol. Ecol. Resour. 22, 2732–2743 (2022).
Rodriguez, M. & Makałowski, W. Software evaluation for de novo detection of transposons. Mobile DNA 13, 1–14 (2022).
Riehl, K. et al. TransposonUltimate: software for transposon classification, annotation and detection. Nucleic Acids Res. 50, e64–e64 (2022).
Bell, E. A. et al. Transposable element annotation in non model species: the benefits of species specific repeat libraries using semi automated EDTA and DeepTE de novo pipelines. Mol. Ecol. Resour. 22, 823–833 (2022).
Faulk, C. De novo sequencing, diploid assembly, and annotation of the black carpenter ant, Camponotus pennsylvanicus, and its symbionts by one person for $1000, using nanopore sequencing. Nucleic Acids Res. 51, 17–28 (2023).
Zhang, X., Zhang, R. & Yu, J. New Understanding of the Relevant Role of LINE-1 Retrotransposition in Human Disease and Immune Modulation. Front. Cell Dev. Biol. 8, 657 (2020).
Acknowledgements
This work was supported by the King Abdullah University of Science and Technology (KAUST) Office of Sponsored Research (OSR) under award numbers FCC/1/1976-44-01, FCC/1/1976-45-01, REI/1/5202-01-01, REI/1/4940-01-01, and RGC/3/4816-01-01, and the National Natural Science Foundation of China under Grant: No.62002388.
Author information
Authors and Affiliations
Contributions
X.L., W.Z., and J.Z. researched the literature. X.L., W.Z., J.Z., H.L., X.X., B.Z., and X.G. contributed substantially to discussions of the content. X.L. wrote the paper, and X.G. reviewed and edited the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Indranil Malik and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: George Inglis.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liao, X., Zhu, W., Zhou, J. et al. Repetitive DNA sequence detection and its role in the human genome. Commun Biol 6, 954 (2023). https://doi.org/10.1038/s42003-023-05322-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-05322-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
