
Abstract
Purpose
Single-nucleotide polymorphism (SNP) microarrays can easily identify whole-chromosome isodisomy but are unable to detect whole-chromosome heterodisomy. However, most cases of uniparental disomy (UPD) involve combinations of heterodisomy and isodisomy, visualized on SNP microarrays as long continuous stretches of homozygosity (LCSH). LCSH raise suspicion for, but are not diagnostic of, UPD, and reporting necessitates confirmatory testing. The goal of this study was to define optimal LCSH reporting standards.
Methods
Eighty-nine individuals with known UPD were analyzed using chromosomal microarray. The LCSH patterns were compared with those in a phenotypically normal population to predict the clinical impact of various reporting thresholds. False-positive and -negative rates were calculated at various LCSH thresholds.
Results
Twenty-seven of 84 cases with UPD had no significant LCSH on the involved chromosome. Fifty UPD-positive samples had LCSH of varying sizes: the average size of terminal LCSH was 11.0 megabases while the average size of interstitial LCSH was 24.1 megabases. LCSH in the normal population tended to be much smaller (average 4.3 megabases) and almost exclusively interstitial; however, overlap between the populations was noted.
Conclusion
We hope that this work will aid clinical laboratories in the recognition and reporting of LCSH.
Similar content being viewed by others


Introduction
Uniparental disomy (UPD) is defined as the inheritance of both homologs of a whole chromosome or chromosome segment from only one parent. There are many mechanisms that can result in UPD, with trisomy rescue being the most common.1 UPD is a rare phenomenon but has serious clinical implications when it occurs either for an imprinted chromosome or, in the case of isodisomy, when one of the parents is a carrier for a recessive disorder.2
Genetic testing for UPD can be performed by a number of molecular techniques, most commonly either microsatellite testing (genotyping highly polymorphic microsatellites on the chromosome of interest in the proband and parents to determine the inheritance of the chromosome of interest) or methylation studies of imprinted loci via methods such as Southern blot, methylation specific (bisulfite) polymerase chain reaction, or methylation-sensitive multiplex ligation-dependent probe amplification (MS-MLPA). However, with the increasing utilization of single-nucleotide polymorphism (SNP) microarrays, it has become clear that blocks of long continuous stretches of homozygosity (LCSH) suggestive of UPD can be detected by examining the allele patterns across various chromosomes. Whole-chromosome isodisomy is easily observed by SNP microarray due to the complete absence of heterozygosity along the entire length of a chromosome. However, whole-chromosome isodisomy represents a minority of all UPD cases. Whole-chromosome heterodisomy, which is more common, is not detectable due to complete lack of LCSH. However, in many cases of UPD there is a mixture of heterozygosity and homozygosity along the length of the affected chromosomes that are generated by crossovers during meiosis in a parental gamete. On a SNP microarray, this is represented visually as LCSH spanning a portion of the chromosome. Such regions indicate potential UPD; however, they also occur due to chance or identity by descent. Therefore, the observation of LCSH on a SNP microarray is not diagnostic of UPD and must be confirmed using other methods.3
The goal of this study was to examine the patterns of LCSH in patients with known UPD. This data was then used to determine optimal thresholds for reporting LCSH observed by microarray to maximize the number of cases of UPD detected and minimize the number of false positives. To do this, 89 individuals with UPD as demonstrated by either microsatellite genotyping or MS-MLPA were analyzed. Peripheral blood DNA from these patients was analyzed using a copy number plus SNP chromosomal microarray to analyze the size and distribution of LCSH observed on the given chromosome with UPD. These were compared with the sizes of LCSH in a phenotypically normal population to establish false-positive and false-negative rates at various thresholds for reporting LCSH.
Materials and methods
This study was approved by the Mayo Clinic Institutional Review Board (IRB 13-003298). A total of 31 individuals were identified with known UPD by microsatellite genotyping. These individuals had UPD for chromosomes 7 (n = 4), 14 (n = 1), 15 (n = 24), and 16 (n = 2). An additional 58 individuals were identified with Prader–Willi syndrome (PWS), diagnosed by MS-MLPA, that was not due to a deletion and therefore very likely due to UPD.4 A population of healthy individuals was also studied in order to estimate the size of LCSH in the absence of uniparental disomy. A total of 3,056 genomic DNA samples from phenotypically normal individuals that comprise the Affymetrix Database of Variants in phenotypically healthy individuals were initially ascertained. Any LCSH >1 megabase was flagged. Individuals with greater than 5% genome-wide LCSH, indications of abnormal chromosomes, or poor quality control metrics were excluded, leaving 2,703 control samples available for analysis of LCSH. This control set was further restricted to analysis of LCSH on chromosomes 11, 14, and 15 only. The rationale for this restriction was to ensure the absence of UPD, as these three chromosomes are the only known chromosomes for which there is a clinically recognizable syndrome resulting from both maternal and paternal UPD.
Microsatellite analysis was performed on a subset of patients by genotyping markers on the chromosome for which UPD was questioned by the referring physician: chromosome 7: D7S515, D7S2423, D7S641, D7S630, D7S510, D7S493, D7S516, D7S530, D7S657, D7S2465, D7S484, D7S517; chromosome 14: D14S276, D14S63, D14S70, D14S283, D14S985, D14S74, D14S275, D14S258, D14S292, D14S288, D14S261; chromosome 15: D15S120, D15S131, D15S165, D15S153, D15S1002, D15S205, D15S127, D15S128, D15S1007, D15S978, D15S1012, D15S130, D15S975, D15S1019, D15S975, D15S1019, D15S986; and chromosome 16: D16S3130, D16S520, D16S521, D16S500, D16S503, D16S516, D16S515, D16S3034, D16S418, D16S3041, D16S505, D16S3057.
The proband and parental samples were genotyped for each marker by polymerase chain reaction amplification using primers flanking the microsatellite (sequences available upon request). One of each primer pair was labeled using a fluorescent dye (FAM, NED, VIC, or HEX). Samples then underwent capillary electrophoresis on an ABI 3100 (Thermo Fischer Scientific, Waltham, MA). Samples were analyzed using GeneMarker (SoftGenetics, State College, PA), and the proband’s genotype for each microsatellite was compared with that of the parents to determine inheritance.
MS-MLPA was performed on a subset of patients using kit ME028-B1 (MRC-Holland, Amsterdam, the Netherlands) according to the manufacturer’s instructions.5 Samples underwent capillary electrophoresis on an ABI 3100, and data was analyzed using GeneMarker. Copy-number probes were analyzed to determine presence/absence of a deletion in the Prader–Willi/Angelman syndrome critical region, and methylation-sensitive probes were analyzed at imprinted loci in order to identify methylation patterns consistent with UPD.
Chromosomal microarray was performed on the CytoScan HD Suite (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. Data was analyzed using ChAS software version 3.1. For automated flagging of LCSH for patients with UPD, a threshold of 3 megabases was used; however, the chromosomes of interest were also manually analyzed to ensure the accuracy of LCSH calls, that LCSH smaller than 3 megabases were recognized, and that for samples positive for UPD15, small deletions within the Prader–Willi/Angelman syndrome critical region, which could potentially be missed by MS-MLPA, were not present. The entire genome was also scanned to ensure that samples with multiple genome-wide LCSH (due to consanguinity) were identified and removed from further analysis.
Results
A total of 89 samples with either confirmed UPD by microsatellite genotyping or likely UPD (methylation-positive, deletion-negative PWS) were analyzed by SNP microarray. Five samples were excluded due to genome-wide LCSH indicative of consanguinity (>2% autosomal homozygosity; one microsatellite positive, four with PWS by MS-MLPA), leaving 84 samples (30 with UPD for various chromosomes confirmed by microsatellite testing; 54 with deletion-negative PWS identified by MS-MLPA) available for further analysis. Of the 30 samples with confirmed UPD, 10 had no LCSH on the chromosome of interest (whole-chromosome heterodisomy), indicating that approximately one-third (33.3%) of all cases of UPD are not detectable by SNP microarray. Of the 54 samples with PWS (deletion-negative, likely UPD), 17 (31.4%) had no LCSH on chromosome 15, indicating that this sample set is similar to the known UPD sample set and that the a non-UPD mechanism resulting in PWS is responsible for few, if any, of these patients (Figure 1). Therefore, the two sets of individuals were combined for all further analyses.

Flow chart illustrating the uniparental disomy (UPD)-positive individuals analyzed in this study. AOH, absence of heterozygosity; LCSH, long continuous stretches of homozygosity; PWS, Prader–Willi syndrome.
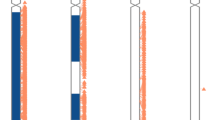
Of the 57 samples remaining (excluding those with no LCSH), 7 (12.3%) had whole-chromosome absence of heterozygosity, consistent with whole-chromosome isodisomy. The remaining 50 (3 with UPD7, 1 with UPD14, and 46 with UPD15) samples had LCSH of variable sizes and locations (Figure 2). Not surprisingly, the interstitial LCSH tend to be much larger than terminal LCSH (average 24.1 Mb versus 11.0 Mb; Figures 2 and 3). The smallest region of interstitial LCSH was a 4.8-megabase region on chromosome 15, while the smallest region of terminal LCSH detected was a 2.9-megabase region, also on chromosome 15.

Relative locations, sizes, and patterns of long continuous stretches of homozygosity (LCSH) in individuals with uniparental disomy (UPD) 15 (proven by microsatellite testing or highly probable by methylation testing),n = 46. LCSH found in the same patient are connected by black dashed lines (LCSH is not drawn to scale). AOH, absence of heterozygosity.

Distribution of sizes of interstitial long continuous stretches of homozygosity (LCSH) (blue) versus terminal LCSH (red) in patients with uniparental disomy (UPD).
The sizes and distribution of LCSH in a phenotypically normal population were also examined. Analysis was restricted to LCSH on chromosomes 11, 14, and 15 only because both maternal and paternal UPD of other chromosomes do not necessarily result in an obvious abnormal phenotype (n = 555 individuals, 615 LCSH identified). As expected, the average size of LCSH in this population was significantly smaller (4.3 megabases) than LCSH representative of UPD (24.1 megabases for interstitial LCSH; Figure 4). Of importance, LCSH in the absence of UPD was almost exclusively interstitial; only one terminal LCSH of 3.6 megabases was identified in a single individual with no obvious evidence of consanguinity.

Comparison of long continuous stretches of homozygosity (LCSH) at various size ranges in individuals with uniparental disomy (UPD) (red) versus individuals without UPD (blue).
Finally, to determine optimal reporting criteria for reporting regions of LCSH detected by SNP microarray, false-negative and false-positive rates at various LCSH reporting thresholds were calculated (Table 1). As there is overlap in the sizes of LCSH between the UPD-positive and UPD-negative individuals, there is no threshold at which sensitivity and specificity are both 100%. As expected, as the reporting size decreases, sensitivity increases but at the cost of decreasing specificity. This effect is much more dramatic for interstitial LCSH; a reporting threshold of 7.5 or 10 Mb would result in only one false negative. However, the number of false positives reported drops significantly as the size threshold is increased (20 false positives at 7.5 Mb versus 13 false positives at 10 Mb). For terminal LCSH, a threshold as low as 5 Mb would result in several false negatives. Because terminal LCSH is so rare in cases of non-UPD, at any size the false-positive rate should be extremely low. Laboratories may choose to have higher size thresholds (15–20 Mb) for interstitial LCSH on a chromosome for which there is no phenotype associated with UPD to reduce false-positive rates further. Depending on the microarray SNP density and software settings, imprinted chromosomes could also be manually examined for any terminal LCSH; however, if such manual scrutiny is applied, it is important to establish a normal baseline for these observations. As a result of these data, the Mayo Clinic Cytogenetics Laboratory has set thresholds of 10 Mb for interstitial LCSH on imprinted chromosomes and 15 Mb for interstitial LCSH on nonimprinted chromosomes. Our threshold for terminal LCSH is 5 Mb regardless of chromosome; however, imprinted chromosomes are inspected for terminal LCSH if the reason for testing heightens suspicion for a disorder involving an imprinted chromosome (hypotonia in a newborn, for example).
Discussion
Because the recommendation was made that chromosomal microarray should be a first-tier test for individuals with phenotypic features such as developmental delay, congenital anomalies, and autism,6 it has become increasingly utilized in the clinic. At present, most microarrays utilized in the clinical laboratory include SNP genotyping in addition to copy-number detection; therefore they are able to detect LCSH suggestive of potential UPD. However, there are currently no guidelines for clinical laboratories to reference when determining thresholds for reporting LCSH.
To our knowledge, only one previous study has examined LCSH detected by SNP microarray in an attempt to determine reporting thresholds. Papenhausen et al.7 performed a smaller study in which they found, in a retrospective sampling of 9 patients with confirmed UPD, that the smallest LCSH was 13.5 megabases in size, compared with 120 patients referred for SNP microarray testing in which the average size was 3.64 megabases. A prospective study was then performed using thresholds determined from the retrospective study (>13.5 megabases for interstitial LCSH, >5 megabases for terminal LCSH, and >15 megabases for sum of multiple LCSH on a single chromosome) in which 29 of 46 cases (63%) in which parental follow-up was obtained were positive for UPD. Although our study did not include a prospective analysis, the sample size is significantly larger and includes a truly normal population to compare with; therefore, this analysis is likely a more accurate representation of LCSH due to UPD versus a truly negative UPD control group. Our retrospective analysis, using UPD positive samples that were identified irrespective of LCSH, along with the normal control comparison, allows for determination of more accurate false-positive and false-negative rates at various LCSH reporting thresholds.
This study demonstrates several important points. First, it is important to note that SNP-based microarrays will not detect approximately 1/3 of UPD cases due to lack of LCSH (complete heterodisomy). Therefore, if a UPD-related syndrome is part of a patient’s differential diagnosis and a chromosomal microarray is normal, additional testing via microsatellite genotyping or methylation studies should be considered. An additional caveat is that SNP-based microarrays cannot reliably detect potential UPD in the background of consanguinity. Third, the size of interstitial LCSH is significantly larger than that of terminal LCSH when UPD is present, and terminal LCSH are extremely rare in the absence of UPD and consanguinity; therefore it may be useful to evaluate terminal and interstitial LCSH differently.
There are some potential drawbacks to note in our study. First and foremost, the majority of the data are derived from chromosome 15, which may or may not be representative of LCSH patterns on other chromosomes. In addition, a large number of samples examined were obtained from samples determined to be positive for PWS but lacking a deletion. Rare examples exist of patients with PWS who have abnormal methylation patterns in the absence of UPD; however, the likelihood of inclusion of samples with this mechanism in our presumed UPD series is very small.
Finally, each laboratory has to take into consideration the microarray platform in clinical use. There are several commercially available copy-number + SNP whole-genome microarrays at the present time with varying SNP densities; this study was performed using a microarray with high-density SNP coverage. Microarrays with lower SNP density may not detect or size LCSH as accurately as microarrays with higher SNP density and may even have more false-positive LCSH calls.8 Therefore, these thresholds may not be applicable to laboratories with lower SNP density chromosomal microarrays.
In conclusion, this study compared the sizes and distribution of LCSH on chromosomes positive for UPD versus LCSH on chromosomes found in phenotypically normal individuals in the absence of UPD. A reporting threshold resulting in 100% sensitivity and specificity is not attainable; however, our data support the utilization of higher size thresholds for reporting interstitial LCSH than for terminal LCSH when using a high-density SNP microarray.
References
Gardner R & Grant R. Chromosome Abnormalities and Genetic Counseling, 3rd edn, Oxford University Press: New York, 2004.
Wang J-C. Genomic imprinting and uniparental disomy. In: Gersen SL, Keagle MB (eds). The Principles of Clinical Cytogenetics. Springer: New York, 2013: 480–489.
Kearney HM, Kearney JB & Conlin LK. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med 2011;31:595–613, ix.
Driscoll DJ, Miller JL, Schwartz S & Cassidy SB. Prader-Willi syndrome. In: Pagon RA, Bird TD, Dolan CR, Stephens K (eds). GeneReviews. University of Washington: Seattle,: WA, 1993.
Procter M, Chou LS, Tang W, Jama M & Mao R. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin Chem 2006;52:1276–83.
Miller DT, Adam MP, Aradhya S et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749–64.
Papenhausen P, Schwartz S, Risheg H et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am J Med Genet A 2011;155A:757–68.
Mason-Suares H, Kim W, Grimmett L et al. Density matters: comparison of array platforms for detection of copy-number variation and copy-neutral abnormalities. Genet Med 2013;15:706–12.
Acknowledgments
Thermo Fisher Scientific provided the Affymetrix CytoScan HD Suite microarrays used in this study. We thank the individuals whose samples were utilized for this study. We also thank Juan Cuevas, Linda Baughn, and Umut Aypar for their helpful suggestions and comments regarding the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Hoppman, N., Rumilla, K., Lauer, E. et al. Patterns of homozygosity in patients with uniparental disomy: detection rate and suggested reporting thresholds for SNP microarrays. Genet Med 20, 1522–1527 (2018). https://doi.org/10.1038/gim.2018.24
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2018.24
Keywords
This article is cited by
-
Chromosomal microarray analysis for prenatal diagnosis of uniparental disomy: a retrospective study
Molecular Cytogenetics (2024)
-
Genetic testing for fetal loss of heterozygosity using single nucleotide polymorphism array and whole-exome sequencing
Scientific Reports (2024)
-
Contribution of uniparental disomy to fetal growth restriction: a whole-exome sequencing series in a prenatal setting
Scientific Reports (2024)
-
Uniparental disomy is a chromosomic disorder in the first place
Molecular Cytogenetics (2022)
-
Prenatal diagnosis of fetuses with region of homozygosity detected by single nucleotide polymorphism array: a retrospective cohort study
Journal of Human Genetics (2022)
