
Abstract
Purpose
Uniparental disomy (UPD) is the rare occurrence of two homologous chromosomes originating from the same parent and is typically identified by marker analysis or single-nucleotide polymorphism (SNP)-based microarrays. UPDs may lead to disease due to imprinting effects, underlying homozygous pathogenic variants, or low-level mosaic aneuploidies. In this study we detected clinically relevant UPD events in both trio and single exome sequencing (ES) data.
Methods
UPD was detected by applying a method based on Mendelian inheritance errors to a cohort of 4912 ES trios (all UPD types) and by using median absolute deviation–scaled regions of homozygosity to a cohort of 29,723 single ES samples (isodisomy only).
Results
As positive controls, we accurately identified three mixed UPD, three isodisomy, as well as two segmental UPD events that were all previously reported by SNP-based microarrays. In addition, we identified three segmental UPD and 11 isodisomy events. This resulted in a novel diagnosis based on imprinting for one patient, and adjusted genetic counseling for another patient.
Conclusion
UPD can easily be identified using both single and trio ES and may be clinically relevant to patients. UPD analysis should become routine in clinical ES, because it increases the diagnostic yield and could affect genetic counseling.
Similar content being viewed by others

INTRODUCTION
Uniparental disomy (UPD) is a copy-neutral chromosome variation defined as the occurrence of two homologous chromosomes, or segments of chromosomes, originating from the same parent without contribution of the other parent.1 Two different types of UPD are generally distinguished: uniparental isodisomy (iUPD), where a single parental homolog is transmitted in duplicate, resulting in whole chromosomal homozygosity (two identical chromosomes); and uniparental heterodisomy (hUPD), in which case the full pair of chromosome homologs is transmitted from a single parent, often after a trisomy rescue. Due to meiotic recombination, a UPD often contains both isodisomic and heterodisomic parts, with either terminal region of homozygosity (ROH) (meiosis I error) or centromeric ROH (meiosis II error), which we refer to as mixed UPD (mixUPD). Segmental UPD may occur after a cell has rescued a pathogenic variant or chromosomal imbalance during mitotic recombination (segUPD). As a result, the isodisomic part of the chromosome pair originates from one parent, whereas the remainder is of biparental origin.2
UPDs do not necessarily have a pathogenic effect as there is no loss or gain of chromosomal material. However, particular events may lead to disease due to imprinting effects. In addition, iUPDs, characterized by a complete homozygous chromosome, may encompass one or more homozygous, pathogenic variants of which the parent of origin is a heterozygous carrier. Finally, UPDs could indicate other chromosomal aberrations such as low-level mosaic aneuploidies, as a result of incomplete rescue, that may contribute to disease. As such, the presence and therefore the detection of UPDs can be medically relevant, and increase the diagnostic yield of genetic testing as well as affect the genetic counseling of patients.3
Over the past few years, cases of UPD were detected by either targeted testing as short tandem repeat (STR) analysis or, in many cases, with SNP-based microarray testing. With the increased use of microarray diagnostics as the main genetic test for patients with neurodevelopmental disorders and/or congenital anomalies, the detection of UPDs expanded. Cases of UPD can be identified either by the detection of a large ROH in single cases, or by checking Mendelian inheritance errors (MIE) in SNP-based microarray data of patient–parent trios.4,5
In recent years genetic laboratories have increasingly applied next-generation sequencing–based technology for the diagnosis of patients with rare disease. In particular, exome sequencing (ES) has become a routine diagnostic investigation.6 Although it is possible to identify UPDs in ES data, these events are not routinely detected as part of a genetic diagnostic analysis strategy. Similarly to genomic microarrays, iUPDs can be detected relatively straightforwardly by identifying homozygosity for a single chromosome. Several algorithms to identify these events have already been published,7,8 but so far there are no reports on their routine application for the detection of iUPD events.9 hUPD cannot be identified based on ES data from a single patient only. However, a trio approach where a patient as well as the parents are sequenced is common, especially for neurodevelopmental disorders. In such cases, genotyping data can be used to detect MIE that are indicative for UPD events. Here we apply both approaches to a large cohort of patients and patient–parent trios for whom diagnostic ES was performed. We aimed to investigate the sensitivity of these approaches and the prevalence of medically relevant UPD events in this patient cohort.
MATERIALS AND METHODS
Samples
We used a de-identified ES cohort of 29,723 individuals, of whom 4912 patients were sequenced in a patient–parent trio approach that we refer to as the full cohort. These data were gathered as part of a routine genetic investigation from Genome Diagnostics Nijmegen, representing patients with a wide variety of different clinical diagnoses (https://gdnm.nl/).10 ES data were analyzed as previously reported.11 Patient samples, together with a basic phenotype description and molecular diagnosis (when available), were anonymized.
Validation data set
To validate our approach we selected eight cases with previously identified UPD events based on trio SNP-based microarrays and where ES data were available (Table 1). These samples were included as a part of the full cohort.
Analysis of single cases
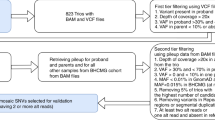
For single patients for whom there were no parental ES available, we identified ROH using the H3M2 algorithm v.2016.10.13 (https://sourceforge.net/projects/h3m2/) that uses BAM files as input.7 We applied a median absolute deviation (MAD) to scale the total ROH size per chromosome across the entire cohort using the robust scale function from quantable package v0.3.6 on R v.3.5.1 (https://cran.r-project.org/package = quantable). We excluded samples with large ROHs (MAD > 3) on more than two chromosomes, as such events are most likely due to consanguinity or identity by descent rather than UPD. After verification of log transformed chromosomal ROH sizes normal distribution (Supplementary Figs. 1, 2), we processed the resulting MAD scores into statistical p values. P values were corrected using the Bonferroni method based on the number of tested chromosomes (n = 653,906). We manually verified events with the most significant p values (–log10 p value = 100, Supplementary Table 2) by inspecting the homozygosity plots generated by H3M2.
Analysis of ES trios
For ES trio analysis, we used UPDio with VCF input file to detect UPD events based on MIE. We applied UPDio v1.0 with fixed plot script.12 UPDio detects uniparental disomy from child–mother–father genotype data with a binomial test to identify chromosomes with a significant burden of uniparentally inherited genotypes and performs multiple testing correction based on the number of tests. Because copy-number variations (CNVs) can affect the identification of UPD events by giving rise to false positive MIE, we removed variants in regions with common CNVs (>1% population frequency, provided by UPDio) and regions called as a CNV within the individual sample. We detected CNVs for all samples using the Conifer13 algorithm as reported previously.14 P values were corrected by the Bonferroni method based on the number of samples (n = 4912) and we selected samples with –log10 p value higher than 48 according to the p value distribution (Fig. 1a).

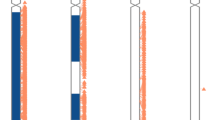
(a) Distribution of –log10 p value from region of homozygosity (ROH) single exome sequencing (ES) analysis and Mendelian inheritance error (MIE) trio ES analysis. Dashed red line = significant threshold. (b) Distribution of uniparental disomy (UPD) calls of the cohort across the genome. Each line is a sample. Blue: isodisomic part, orange: heterodisomic part. *Segmental UPD is confirmed for 4 cases and is suspected for 1 case.
RESULTS
UPDive calls
Among the 4912 ES trios, we identified 10 (0.2%) UPD events, 4 of which involved iUPD (on chromosomes 1, 4, 7, and 10), 3 mixUPD (on chromosomes 2, 16, and 22), and 3 segUPD (on chromosomes 3, 8, and 13) (Supplementary Fig. 3). For single analysis, we selected samples with –log10 p value = 100 according to p value distribution. In the cohort of 29,723 single cases (including the patients of the 4912 trios), we found 14 (0.05%) cases with isodisomy (3 of chromosome 1; 2 of chromosome 19; 1 of chromosomes 2, 4, 7, 8, 10, 12, 15, 20, and 22) and 2 significant ROH on a single chromosome (chromosomes 11 and 15) (Fig. 1b, Supplementary Fig. 4).
Performance on validation samples
To validate our methods, we initially selected eight different UPDs (three mixUPD, three iUPD, and two segUPD) that were previously identified by SNP-based microarrays and available in our ES cohort. For seven of the eight events parental data (trios) were available (Table 1, Supplementary Table 1). Trio ES analysis successfully detected all UPD events (7/7). Our single ES approach successfully identified three of three iUPD using only single cases, and two of two segUPD but with low p value.
Novel UPD events detected in full cohort
In total, we detected 14 events suggestive of UPDs that were not part of our validation set, due to either isodisomy (n = 11) or the presence of a unique large ROH (n = 3) ranging in size from 67 Mb to 100 Mb, highly suggestive for UPD. For the 11 suggestive iUPDs the entire chromosome of interest was homozygous and there were no significant additional ROH on other chromosomes, and therefore it is very likely that these events are iUPD. For the three cases with a large ROH, we could confirm the segUPD based on the independent trio ES analysis (–log10 p value N9 = 50 and N10 = 5, confirmed by plot visualization) for two samples (Supplementary Fig. 6). For the other case, no validation was possible due to lack of parental samples. For four of the novel cases, it turned out that a UPD was previously reported based on other tests, but this was unknown to us at the time of analysis (Table 1, final column). For the remaining cases methylation or STR analysis of the patient sample could possibly confirm these events.15
Diagnostic implications
For all 14 patients with novel UPD events we reevaluated the existing ES analysis. For three cases, the detected UPD events would give rise to an imprinting disorder (Table 1; cases N5, N10, and monozygotic twins N11 and N12). For example, in case N5, the maternal isodisomy of chromosome 7 would lead to Silver–Russell syndrome (OMIM 180860), a genetic diagnosis that fits with the clinical phenotype of this patient. In two cases a homozygous pathogenic variant was identified in the UPD chromosome, meaning that only a single parent would be a heterozygous carrier of the respective variant. This changed the genetic counseling for their families, for whom there is almost no recurrence risk in this case, compared with the autosomal recessive recurrence risk of 25%. This concerned a homozygous nonsense variant, Chr13(GRCh37):g.103514595C>T; NM_000123.3:c.1096C>T (p.(Arg366*), in the ERCC5 gene (case N9), which gives rise to a genetic diagnosis of cerebrooculofacioskeletal syndrome type 3 (OMIM 616570), which matched with the phenotype of the patient. In case N14 the well-known tumor risk–associated 1100delC variant (Chr22[GRCh37]:g.29091857del; NM_007194.3:c.1100del; p.[Thr367fs]) in the CHEK2 gene was detected in the homozygous state in a patient with a history of multiple primary tumors that would fit with a clinical diagnosis of the CHEK2-related cancer susceptibility (OMIM 609265). UPD identification is of unknown significance (UUS) for the remaining eight patients.
DISCUSSION
In this study, we report a robust and validated UPD detection method on a large ES data set. The UPD prevalence is 0.2% in our cohort of 4912 trio ES and the iUPD prevalence is 0.05% in our single ES cohort of 27,923 samples. This is concordant with previous studies based on SNP-based microarray16 and estimates of UPD in the general population.17 In 9/22 of cases the detection of the UPD is of direct diagnostic relevance and affects genetic counseling because of altered recurrence risk.
In a relatively high percentage of cases (12/22 = 54%) the identified UPD is a UUS. This is partly due to the retrospective nature of our study, where we are unable to perform additional investigations. If the detection of UPD would have been performed as part of the routine investigation, follow-up studies would have allowed us to determine the clinical significance of a larger fraction of these UPD events, increasing the overall diagnostic yield of ES. Follow-up analysis with methylation or STR analysis could confirm UPD and cytogenetic testing such as karyotyping and/or fluorescence in situ hybridization (FISH) analysis may lead to the detection of mosaic aneuploidies, or an abnormal, balanced karyotype.3 Moreover, disease genes on the UPD chromosome that fit well with the clinical phenotype of the patient could be analyzed in more detail. For example, case N1 (UPD[1]) of our study is a patient suspected of having Stargardt disease, for which the gene ABCA4 in 1p22.1 is a known disease gene. For this case, Sanger sequencing of the coding regions of ABCA4 was previously performed, without resulting in a genetic diagnosis. However, with the knowledge on UPD(1) in this case, an in-detail analysis of the ABCA4 gene, including the noncoding regions, would be an obvious next step.
The detection rate of UPD in a single ES analysis depends on the size of the ROH and is lower than many state-of-the-art SNP array platforms that often interrogate a much higher number of SNPs than are obtained from regular ES data. Terminal ROH or centromeric ROH is frequently observed in cases with UPD, but is rare in cases of non-UPD.4 Therefore, the detection rate of UPD in a single ES analysis could be further improved by not only looking at the size, but also at the location of the detected ROH, differentiating between terminal, interstitial, and centromeric. SegUPD has often been reported as mosaic,1 which further challenges the reliable detection of such an event for both SNP arrays as well as ES. In this study, we did not detect a complete heterodisomy in the 4912 trios analyzed. Although complete heterodisomy has been reported in up to 1/3 of patients with an imprinting disorder,4 such a finding is very rare in an unselected patient cohort.
In conclusion, we show how to identify UPD events from exome sequencing data, an approach that can be easily adopted by other clinical laboratories. Detecting UPD events in ES data will increase the diagnostic yield and may have significant implications for the interpretation of ES data, and affect the genetic counseling of recurrence risk. Therefore we believe that UPD analysis should become a routine part of the analysis of ES data in genetic testing laboratories.
Code availability
UPDio v1.0 with fixed plot script available at https://github.com/kyauy/UPDio
All scripts used for the UPDive study are available at https://github.com/kyauy/UPDive.
References
Eggermann T, Soellner L, Buiting K, Kotzot D. Mosaicism and uniparental disomy in prenatal diagnosis. Trends Mol Med. 2015;21:77–87.
Waggoner D, Wain KE, Dubuc AM, et al. Yield of additional genetic testing after chromosomal microarray for diagnosis of neurodevelopmental disability and congenital anomalies: a clinical practice resource of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2018;20:1105–1113.
Liehr T. Uniparental disomy (UPD) in clinical genetics. Berlin: Springer; 2014.
Hoppman N, Rumilla K, Lauer E, et al. Patterns of homozygosity in patients with uniparental disomy: detection rate and suggested reporting thresholds for SNP microarrays. Genet Med. 2018;20:1522–1527.
Ting JC, Roberson EDO, Miller ND, et al. Visualization of uniparental inheritance, Mendelian inconsistencies, deletions, and parent of origin effects in single nucleotide polymorphism trio data with SNPtrio. Hum Mutat. 2007;28:1225–1235.
Adams DR, Eng CM. Next-generation sequencing to diagnose suspected genetic disorders. N Engl J Med. 2018;379:1353–1362.
Magi A, Tattini L, Palombo F, et al. H3M2: detection of runs of homozygosity from whole-exome sequencing data. Bioinformatics. 2014;30:2852–2859.
Narasimhan V, Danecek P, Scally A, et al. BCFtools/RoH: a hidden Markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics. 2016;32:1749–1751.
Erger F, Burau K, Elsässer M, et al. Uniparental isodisomy as a cause of recessive Mendelian disease: a diagnostic pitfall with a quick and easy solution in medium/large NGS analyses. Eur J Hum Genet. 2018;26:1392–1395.
Neveling K, Feenstra I, Gilissen C, et al. A post-hoc comparison of the utility of Sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. 2013;34:1721–1726.
Lelieveld SH, Reijnders MRF, Pfundt R, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–1196.
King DA, Fitzgerald TW, Miller R, et al. A novel method for detecting uniparental disomy from trio genotypes identifies a significant excess in children with developmental disorders. Genome Res. 2014;24:673–687.
Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525–1532.
Pfundt R, Del Rosario M, Vissers LELM, et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genet Med. 2017;19:667–675.
Sharp AJ, Migliavacca E, Dupre Y, et al. Methylation profiling in individuals with uniparental disomy identifies novel differentially methylated regions on chromosome 15. Genome Res. 2010;20:1271–1278.
Wang J-C, Ross L, Mahon LW, et al. Regions of homozygosity identified by oligonucleotide SNP arrays: evaluating the incidence and clinical utility. Eur J Hum Genet. 2015;23:663–671.
Nakka P, Smith SP, O’Donnell-Luria AH, et al. Characterization of prevalence and health consequences of uniparental disomy in four million individuals from the general population. Am J Hum Genet. 2019;105:921–932
Acknowledgements
We sincerely thank all patients, clinicians, biologists, and bioinformaticians involved in this project. This work is generated within the European Reference Network on Rare Congenital Malformations and Rare Intellectual Disability (ERN-ITHACA). This project was in part supported by the Solve-RD project that has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement number 779257.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Yauy, K., de Leeuw, N., Yntema, H.G. et al. Accurate detection of clinically relevant uniparental disomy from exome sequencing data. Genet Med 22, 803–808 (2020). https://doi.org/10.1038/s41436-019-0704-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-019-0704-x
Keywords
This article is cited by
-
Contribution of uniparental disomy to fetal growth restriction: a whole-exome sequencing series in a prenatal setting
Scientific Reports (2024)
-
Autosomal recessive congenital cataract is associated with a novel 4-bp splicing deletion mutation in a novel C10orf71 human gene
Human Genomics (2023)
-
Systematic analysis of paralogous regions in 41,755 exomes uncovers clinically relevant variation
Nature Communications (2023)
-
Exome sequencing for structurally normal fetuses—yields and ethical issues
European Journal of Human Genetics (2023)
-
Functional Characterization of a Novel SLC4A4 Variant and Uniparental Isodisomy in Proximal Renal Tubular Acidosis Patient
Biochemical Genetics (2023)

{kind=link}
{kind=link}
{kind=link}
{kind=link}