
Abstract
Purpose:
Single-nucleotide polymorphism microarray analysis identifies copy-number variants and blocks of homozygosity, suggestive of consanguinity or uniparental disomy. The purpose of this study was to validate chromosomal microarray analysis for the identification of uniparental disomy in a clinical laboratory.
Methods:
In phase I of this retrospective study, nine cases with uniparental disomy for chromosomes 7 (n = 1), 14 (n = 1), and 15 (n = 7), identified by conventional polymorphic microsatellite marker analysis were analyzed on the Affymetrix 6.0 single-nucleotide polymorphism array. In phase II, four cases of uniparental disomy 15 showing heterozygosity for all microsatellite markers were analyzed using the same array.
Results:
Chromosomal microarray analysis detected blocks of homozygosity in eight of the nine cases in phase I. Phase II analysis of molecularly defined heterodisomy failed to detect blocks of homozygosity in three of the four cases. The four cases in which microarray did not detect blocks of homozygosity all involved chromosome 15.
Conclusion:
A failure to recombine may predispose to nondisjunction and, therefore, to uniparental disomy. Four cases of heterodisomy 15 were not detected by array, suggesting a lack of recombination. Therefore, a normal chromosomal microarray result for chromosome 15 does not exclude the possibility of uniparental disomy. This observation may apply to other chromosomes; however, further study is needed.
Genet Med 2012:14(8):753–756
Similar content being viewed by others


Introduction
Uniparental disomy (UPD) is the inheritance of both homologs of a chromosome pair from a single parent with no representative copy inherited from the other parent. Generally, the inheritance of both chromosomes from a single parent is of no clinical consequence, unless it unmasks mutations for an autosomal recessive disease, or if the chromosome contains imprinted genes that have different expression patterns depending on the parent of origin. Several chromosomes (e.g., chromosomes 6, 7, 11, 14, and 15) have clusters of imprinted genes associated with recognizable syndromes and are, therefore, sensitive to UPD; developmental delay is a feature of most of these syndromes. HeteroUPD is the presence of one of each homolog from one parental pair in a diploid zygote, and isoUPD is the presence of one parental homolog in duplicate in a diploid cell. Due to meiotic crossing over, partial heterodisomy and partial isodisomy (iso/heteroUPD) may coexist for the same chromosome pair.
Mechanisms leading to UPD include: (i) trisomy rescue: the fertilization of an abnormal disomic gamete by a normal haploid gamete followed by postzygotic loss of the chromosome derived from the haploid gamete; (ii) gamete complementation: the fertilization of an abnormal disomic gamete by an abnormal gamete nullisomic for the same chromosome; (iii) postfertilization error: a loss of one chromosome followed by duplication of the remaining homolog, and (iv) monosomy rescue: the fertilization of a normal haploid gamete by an abnormal nullisomic gamete and somatic reduplication of the monosomic chromosome.1 Meiotic nondisjunction followed by trisomy rescue in the zygote is a frequent cause of UPD, particularly for maternal nondisjunction of chromosome 15 (Prader–Willi syndrome (PWS), OMIM no. 176270).2 Two well-known factors that predispose to nondisjunction are maternal age3 and aberrant recombination4 (e.g., lack of recombination or recombination too close to the centromere or telomere).
Chromosomal microarray analysis (CMA) is recommended as a first-tier test for the detection of copy-number variants in individuals with developmental delay and autism,5 replacing karyoptype analysis and some disease-specific tests. Single-nucleotide polymorphisms (SNPs) are biallelic markers found throughout the genome, and a SNP-based array interrogates SNP allele status across hundreds of thousands of SNPs throughout the genome. An individual could show homozygosity for a particular SNP because both parents have the same allele. Small blocks of homozygosity are present on multiple chromosomes in all outbred populations.6 However, when large blocks of homozygosity are present on multiple chromosomes it typically represents parental relatedness.7 Alternatively, when large block(s) of homozygosity are restricted to a single chromosome it can involve the whole chromosome and reflect complete isodisomy or it can be interspersed with regions of heterodisomy (iso/heteroUPD). The power of CMA for the detection of copy-number variants has been well documented; however, very few studies have established the clinical use of SNP-based CMA for the detection of UPD.8,9
Methods
UPD was investigated by conventional polymorphic microsatellite analysis after an abnormal methylation test result and the absence of a common microdeletion on chromosome 15, or because of a phenotype suggestive of UPD7 or UPD14. Microsatellite analysis was performed by polymerase chain reaction amplification on samples from the patient and both parents; all cases required at least three microsatellite markers consistent with failure to inherit a parental allele to be considered positive for UPD.
There were two phases to the project. In the first phase, we retrospectively identified all UPD-positive cases identified by polymorphic marker analysis over the time frame of 2007–2011 (N = 9) for CMA. The polymorphic markers used to assess UPD were the ABI HD5 v2.5 linkage mapping panel 52 (D7S2464, D7S644, D7S2252, D7S2427, D7S506, D7S2476, D7S2459, and D7S483; Life Technologies, Carlsbad, CA), the ABI HD5 v2.5 linkage mapping panel 70 (D15S986, D15S118, D15S1016, D15S1036, D15S988, and D15D1014; Life Technologies) and for chromosome 14 (D14S72, D14S50, D14S283, D14S67, and D14S250; primer sequences available on the UCSC Genome Browser, http://genome.ucsc.edu/). The UPD-positive samples were run on the Affymetrix 6.0 array (Santa Clara, CA) and analyzed without the researcher who analyzed the results having prior knowledge of the chromosome involved.
For the second phase of the project, we retrospectively selected all UPD cases identified over the time frame of 1998–2006 (N = 4) for which all microsatellite markers on the patient were heterozygous (i.e., within the limitations of marker analysis, were consistent with complete heteroUPD). These cases would either have complete heterodisomy (i.e., no observable recombinants) or have regions of isodisomy not detected with the microsatellite markers used for analysis. Due to the high prevalence of UPD in PWS, all available cases in the second phase were UPD15. The markers used to assess UPD in these older cases were D15S18, D15S11, D15S113, GABRB3, GABRA5, D15S217, D15S219, IPM15M9, and D15S87 (primer sequences available on the UCSC Genome Browser, http://genome.ucsc.edu/).
All UPD probands were analyzed on the Affymetrix 6.0 SNP array (Santa Clara, CA). Sample preparation and hybridizations were performed according to the manufacturer’s protocols and analyzed using GTC v4.0 and Chromosomal Analysis Suite v1.1 without the analyzer having prior knowledge of the chromosome involved. Human genome build 18 (Hg 18) was used for the analysis. Blocks of homozygosity were visualized when >1 Mb; these were considered suggestive of UPD when the block(s) of homozygosity on a single chromosome were larger than the average blocks of homozygosity throughout the whole genome.
Results
Phase I
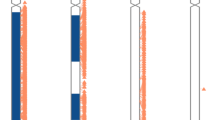
Eight of the nine UPD cases, selected without bias for type of UPD, were detected by CMA (89%; Table 1 ). The confirmed cases consisted of one case of complete isoUPD for chromosomes 14 and 15, as well as five cases of iso/heteroUPD15 and one case of iso/heteroUPD7 detected by CMA as a large block(s) of homozygosity on a single chromosome. The microsatellite markers analyzed for each case were concordant with the array results ( Figure 1 ). One case, which showed heterozygosity for all maternal microsatellite markers on chromosome 15, showed no large block(s) of homozygosity restricted to a single chromosome on CMA. This case was considered to be complete heteroUPD15, with no observable recombinants.

Chromosome 15 ideograms for 6 of the 13 UPD cases studied. (a) Microsatellite analysis shows segments of isodisomy (Iso), and heterodisomy (Het), concordant with CMA. (b) Microsatellite analysis shows heterodisomy; CMA analysis detects heterodisomy in these same regions, and isodisomy in regions not assessed by microsatellite analysis. (c–f) All microsatellite markers analyzed show heterodisomy. No large blocks of homozygosity observed, failing to detect UPD with CMA. The average size of the blocks of homozygosity in these cases was 1.3 Mb. CMA, chromosomal microarray analysis; Mat, maternal; UPD, uniparental disomy.
Phase II
CMA was able to detect UPD15 in only one of the four cases (25%) ( Table 1 ). Three of the UPD cases showed no large block(s) of homozygosity ( Figure 1 ).
Summary
The average size of the blocks of homozygosity across UPD chromosomes detected by CMA was 23.3 Mb (ranging from 13 Mb to 33.7 Mb). The average size of the blocks of homozygosity across the remainder of the autosomes was 1.3 Mb (ranging from 1.2 to 3.7 Mb). The average size of the blocks of homozygosity for the four cases of UPD15 not detected by CMA was 1.3 Mb (ranging from 1.1 to 4.0 Mb) for all autosomes.
Discussion
The utility of CMA in detecting copy-number variation is well documented and is now considered routine clinical practice. The purpose of this study was to validate the use of the Affymetrix 6.0 SNP array for UPD analysis. SNP-based CMAs can identify blocks of homozygosity and are suggestive of UPD when these large blocks are restricted to a single chromosome. In the first phase of the project, we performed CMA on nine UPD cases, of which one case of maternal UPD15 was not detected. In the second phase of our study, we selected cases showing only heterozygosity for all microsatellite markers; these were all cases of UPD15. CMA failed to detect three of the four cases.
It has previously been demonstrated that a high percentage of cases of PWS are due to meiotic errors resulting in UPD, with 25–29% of all PWS cases caused by UPD; therefore, it was not surprising that the majority of cases in our project were UPD15. One study demonstrated that up to 21% of cases of maternal UPD15 show no evidence of recombination during meiosis I.3
In our laboratory, a total of 14 cases of UPD15 were ascertained from 1998 to 2011, 11 of which were included in this study. The three cases not analyzed by CMA each showed homozygosity for at least three maternal markers; given the concordance of microsatellite markers and CMA results for all analyzed cases, we assume these three cases would be detected by CMA. Therefore, of the 14 cases of UPD15, four (29%) were not detected by CMA. In all four cases, the data were consistent with complete heteroUPD with no observable recombination, and, as such, are assumed to arise as a final result of failure of maternal recombination. The similarity in the percentage of complete heteroUPD cases in this study (29%) to those of earlier recombination studies3 for chromosome 15 supports this assumption. If 21–29% of all UPD15 PWS cases show no evidence of recombination, as many as 8% of PWS cases will be missed with CMA alone. However, it is not clear whether the same proportion of nonrecombinants applies to all other chromosomes. Certainly, there are reports that lack of recombination on chromosome 18 is a rare event,10 and, therefore, the percentage of UPD cases not identified by CMA in this study may not be generalizable to other chromosomes. Further study is necessary to determine the percentage of UPD cases for other chromosomes that may also be missed by CMA, particularly for those chromosomes susceptible to UPD disorders.
A recent study by Papenhausen et al.9 performed a similar UPD validation study on the Affymetrix 6.0 array by retrospectively testing eight cases of UPD; blocks of homozygosity were detected in all eight cases. The smallest single block of homozygosity for a single chromosome observed in these eight cases was 13.5 Mb, and there was one case with two large blocks of homozygosity of 11 Mb on a single chromosome. Therefore, the authors’ threshold for reporting possible UPD was a single block of homozygosity of 13.5 Mb for a single chromosome or when the sum of two or more blocks of homozygosity on a single chromosome was over 15 Mb. Using these size criteria, the authors prospectively analyzed 92 cases for UPD using CMA. Of 46 patients that met the proposed criteria for UPD, only 29 were confirmed; the majority of the confirmed cases (n = 21) were associated with chromosomes known to have imprinted genes (e.g., chromosomes 6, 7, 14, and 15). However, in this prospective review design, and in any prospective SNP array analytic design, complete heteroUPD cases would be missed.
In our series of patients, the smallest single block of homozygosity was 13.0 Mb, smaller than that reported by Papenhausen et al.9 In addition, the average size of the blocks of homozygosity across the remaining chromosomes, except the X chromosome, was 1.3 Mb. This is smaller than the 3.64 Mb previously reported.9 The difference in the size may be a result of our study population having more genetic diversity between parents. This was particularly evident in the prospective analysis of UPD, as Papenhausen et al.9 suggested that identity by decent could explain why only 29 of the 46 cases meeting their criteria for UPD were confirmed.
Another retrospective review reported six patients with microsatellite confirmed UPD analyzed on the Affymetrix GeneChip Human Mapping 10K array.8 In the study, analysis of the proband and both parents allowed comparison of SNP transmissions. They identified UPD in all six patients, including three segmental UPDs (postzygotic events). Studying the parents by microarray would identify all cases of UPD, including complete heteroUPD; however, it is not economically feasible to test both parents for each patient in a clinical laboratory setting.
In conclusion, our study demonstrates that SNP-based CMA is a valuable addition to standard CMA, as it will detect the majority of cases of UPD, including all cases of isoUPD and most cases of iso/heteroUPD. However, it will fail to identify cases of complete heteroUPD, as demonstrated in our series where 29% of cases of UPD15 identified by microsatellite analysis were not detected by CMA. Therefore, a normal SNP-based CMA cannot exclude the diagnosis in patients with a suspected imprinting disorder such as PWS, and if a strong clinical suspicion remains, methylation analysis and/or UPD analysis should be pursued.
Disclosure
The authors declare no conflict of interest.
References
Robinson WP . Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays 2000;22:452–459.
Robinson WP, Barrett IJ, Bernard L, et al. Meiotic origin of trisomy in confined placental mosaicism is correlated with presence of fetal uniparental disomy, high levels of trisomy in trophoblast, and increased risk of fetal intrauterine growth restriction. Am J Hum Genet 1997;60:917–927.
Robinson WP, Kuchinka BD, Bernasconi F, et al. Maternal meiosis I non-disjunction of chromosome 15: dependence of the maternal age effect on level of recombination. Hum Mol Genet 1998;7:1011–1019.
Lorber BJ, Grantham M, Peters J, Willard HF, Hassold TJ . Nondisjunction of chromosome 21: comparisons of cytogenetic and molecular studies of the meiotic stage and parent of origin. Am J Hum Genet 1992;51:1265–1276.
Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010;86:749–764.
Gibson J, Morton NE, Collins A . Extended tracts of homozygosity in outbred human populations. Hum Mol Genet 2006;15:789–795.
Li LH, Ho SF, Chen CH, et al. Long contiguous stretches of homozygosity in the human genome. Hum Mutat 2006;27:1115–1121.
Altug-Teber O, Dufke A, Poths S, et al. A rapid microarray based whole genome analysis for detection of uniparental disomy. Hum Mutat 2005;26:153–159.
Papenhausen P, Schwartz S, Risheg H, et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am J Med Genet A 2011;155A:757–768.
Bugge M, Collins A, Petersen MB, et al. Non-disjunction of chromosome 18. Hum Mol Genet 1998;7:661–669.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tucker, T., Schlade-Bartusiak, K., Eydoux, P. et al. Uniparental disomy: can SNP array data be used for diagnosis?. Genet Med 14, 753–756 (2012). https://doi.org/10.1038/gim.2012.35
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/gim.2012.35
Keywords
This article is cited by
-
Copy neutral absence of heterozygosity on chromosome 15 distal long arm: A surrogate marker for Prader–Willi/Angelman syndromes?
Molecular Cytogenetics (2021)
-
Uniparental isodisomy of chromosome 2 causing MRPL44-related multisystem mitochondrial disease
Molecular Biology Reports (2021)
-
SNP arrays: comparing diagnostic yields for four platforms in children with developmental delay
BMC Medical Genomics (2014)
