
Abstract
Rapid identification of clinically significant variants is key to the successful application of next generation sequencing technologies in clinical practice. The Melbourne Genomics Health Alliance (MGHA) variant prioritization framework employs a gene prioritization index based on clinician-generated a priori gene lists, and a variant prioritization index (VPI) based on rarity, conservation and protein effect. We used data from 80 patients who underwent singleton whole exome sequencing (WES) to test the ability of the framework to rank causative variants highly, and compared it against the performance of other gene and variant prioritization tools. Causative variants were identified in 59 of the patients. Using the MGHA prioritization framework the average rank of the causative variant was 2.24, with 76% ranked as the top priority variant, and 90% ranked within the top five. Using clinician-generated gene lists resulted in ranking causative variants an average of 8.2 positions higher than prioritization based on variant properties alone. This clinically driven prioritization approach significantly outperformed purely computational tools, placing a greater proportion of causative variants top or in the top 5 (permutation P-value=0.001). Clinicians included 40 of the 49 WES diagnoses in their a priori list of differential diagnoses (81%). The lists generated by PhenoTips and Phenomizer contained 14 (29%) and 18 (37%) of these diagnoses respectively. These results highlight the benefits of clinically led variant prioritization in increasing the efficiency of singleton WES data analysis and have important implications for developing models for the funding and delivery of genomic services.
Similar content being viewed by others

Introduction
Whole exome sequencing (WES) is a powerful diagnostic tool in clinical practice, but its utility is limited by the potentially overwhelming scale of the genomic data generated. The efficient identification of clinically significant variants is a key component of the successful implementation of genomic technologies in the clinical setting. The ideal variant prioritization tool or strategy should be able to rank highly the causative variant(s) within a patient data set. Some existing tools such as the CADD1 and Condel2 deleteriousness scores are genotype-driven, providing clinical hypothesis-free variant ranking. Other computational tools such as Exomiser,3 eXtasy,4 Phevor5 and PhenGen6 combine phenotype and variant information. A comparison of the performance of these tools using simulated data reported that computational phenotype analysis substantially improves the performance of exome analysis pipelines, with Exomiser outperforming the other tools evaluated.7 Another commonly used strategy is the use of trio WES, which allows the phasing of variants, and rapid identification of de novo variants. This strategy has been reported to increase diagnostic yield by approximately 10%8, 9 but has the disadvantage of increasing the cost of sequencing compared to singleton WES. In addition, parental samples may not always be available, and trio sequencing may be less effective in consanguineous families.
We have previously described the development and implementation of a filtering and prioritization system designed to highlight the results most likely to be relevant in the clinical setting.10 This system has two components: one is a gene prioritization index (GPI) based on clinician-generated a priori candidate gene list, and the other a variant prioritization index (VPI) combining information on rarity, conservation and protein effect. Here we present an evaluation of this clinically driven variant prioritization framework, and compare it with publically available computational tools for ranking of variants from WES data which utilize phenotypic and variant data (Exomiser) or variant data alone (CADD and Condel). We also evaluate two computational tools, PhenoTips11 (http://phenotips.org) and Phenomizer12 (http://compbio.charite.de/phenomizer/) that use phenotypic information to generate lists of differential diagnoses and candidate genes and compare their performance to that of clinicians.
Subjects and methods
Data set
We used a data set derived from 80 infants who presented with multiple congenital abnormalities and dysmorphic features or other features strongly suggestive of monogenic disorders who underwent singleton WES as a first-tier investigation.13 Each patient’s key features were recorded using Human Phenotype Ontology (HPO) terms at enrolment using PhenoTips, and a phenotype-driven list of candidate genes was generated by their clinician based on the clinical differential diagnosis, and supplemented by literature and database searches. A clinical summary of each case was presented by the referring clinician at a dedicated weekly multidisciplinary meeting. The clinicians involved were seven clinical geneticists, two metabolic physicians and two paediatric neurologists. The multidisciplinary meeting determined patient suitability against the study inclusion criteria, and also refined candidate gene lists as necessary. Singleton WES was performed as previously described.13
The Melbourne Genomics Health Alliance variant prioritization framework
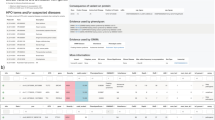
The Melbourne Genomics Health Alliance variant prioritization framework, implemented in the bioinformatics pipeline, Cpipe10 (http://cpipeline.org), has two components. The first is a GPI whereby variants in genes from the clinician-generated candidate gene list were automatically given first-tier priority, while variants in other Online Mendelian Inheritance in Man disease-associated genes (the ‘Mendeliome’) were given second-tier priority. The second component is a VPI that combines factors such as rarity, conservation and predicted effect on the protein structure to rank each variant individually within the data set as well as to place groups of variants in ‘Mendeliome’ genes into four tiers (Figure 1). Cpipe uses ANNOVAR (Version 23 August 2013)14 to identify the functional consequence of variants. Condel scores were generated for each variant using VEP 74, and the 24-1-2013 Condel plugin (incorporating SIFT and PolyPhen-2 scores as predictors). CADD scores were generated for each variant using Annovar version 2013aug23. CADD scores of complex variants were calculated separately using the online application: http://cadd.gs.washington.edu/score.

Variant Priority Index ranks variants based on characteristics such as population frequency, conservation scores and predicted effect on the protein (1000G: 1000 Genomes, ESP: Exome Sequencing Project).
Variants that did not meet the criteria for even the first VPI tier, and those in genes not currently associated with human disease were not considered for curation. Variants that reached these minimum requirements for prioritization were imported into a customized Leiden Open Variation Database15 and sorted by GPI then by VPI (both in descending order) to produce the combined ranking. If no causative variant was identified, the variants in the ‘Mendeliome’ were re-ranked by VPI alone. Variants in the V4 and V3 categories (truncating/splice-site or very rare missense) were reviewed by a clinician to determine whether the affected gene was consistent with the patient phenotype. Variants could also be sorted by any individual attribute, such as GPI, VPI, Condel score or CADD score.
Each patient’s data were analysed individually when available, over a period of 24 months (February 2014 to February 2016). The analysis and annotation pipeline, and the gene content of the ‘Mendeliome’ were updated throughout this time, with data from ‘unsolved’ patients reanalysed on a 6-monthly basis.
Determination of causality and variant classification in all cases was made by consensus at a multidisciplinary team meeting, comprising clinical and laboratory geneticists, bioinformaticians, genetic counsellors and other medical specialists. Criteria for variant classification were based on the principles outlined in the American College of Medical Genetics and Genomics standards for interpretation of sequence variants.16 A diagnosis of 49 genetic conditions was made in 47 out of 80 infants (59%), including one further case since the original publication.13 A total of 59 variants were designated as causative: 33 were truncating, two were splice-site mutations, two were in-frame deletions or duplications, one was a stop loss, and 21 were missense. All novel variants determined to be causative have been deposited in ClinVar.
Comparison with other tools
The WES data from the 80 infants were used to evaluate the performance of the clinically driven variant prioritization framework and to compare it to other publically available computational resources. The full data set was reanalysed to determine the ranking provided by the GPI and VPI tools used independently, and the ranking provided by CADD and Condel scores. The recorded HPO terms and the VCF files were entered in Exomiser between April 2016 and June 2017. The default settings were used, applying a 1% minor allele frequency cutoff and no inheritance model. The human–disease association data analysis model (PhenIX) was used. For the 47 patients with a WES diagnosis, the presence or absence of the causative variant, and its ranking was recorded. The variant rankings produced by Exomiser and CADD scores for all patients including the 33 patients without a WES diagnosis were reviewed by a clinical geneticist for new diagnoses.
We determined the average ranking, and the proportion of causative variants that were ranked top, and within the top 5 variants, by each tool. For 10 of the 47 infants, multiple causative variants were identified due to either the condition being autosomal recessive, or due to the presence of multiple unrelated diagnoses. In order to compare the ranks of all variants across all patients, the ranks of these second and subsequent causal variants were adjusted such that for each ranking method, the adjusted rank was equal to the original rank minus the number of causal variants ranked higher. For example, if the prioritization tool ranked the two causative variants for an autosomal recessive condition as number one and two, they were taken as having both been ranked one. Exomiser ranks multiple variants in the same gene together, and these were treated as a single ranking regardless of the number of variants per gene.
In addition, we evaluated the number of WES diagnoses that were included in a priori lists of differential diagnoses based on patient phenotype by clinicians and by patient phenotyping software. The presence or absence of the WES diagnosis in the list of differentials produced by PhenoTips was audited at the end of the study. The HPO terms recorded by clinicians at enrolment were entered into Phenomizer in February 2016. The presence or absence of the WES diagnosis in the top 100 differentials provided, and its ranking was recorded.
Ethics approval
The study was part of the Melbourne Genomics Health Alliance demonstration project (www.melbournegenomics.org.au), and received Human Research Ethics Committee approval (13/MH/326).
Results
The HPO terms and causative variant rankings for each patient are listed in Supplementary Table 1. Clinicians described patient phenotypes using an average of four positive HPO terms (range 1–12). The median length of clinician-generated gene list was 23 (range 1–161). An average of 107 variants were available for curation in each patient (range 70–168). Of these, an average of 1.4 variants were in genes from the clinician-generated candidate gene list (range 0–8). Thirty-nine out of 49 (79%) of molecular diagnoses made were from the clinician-generated candidate gene lists, and the average rank of the causative variant was 1.09. Of the 14 variants that did not receive a GPI rank because they were outside the gene list, the average rank produced by the VPI was 5.79. Overall, the average rank of causative variants produced by the combined use of GPI and VPI was 2.24 (range 1–14), with 45 (76%) variants ranked first, and 53 (90%) ranked within the top 5 variants in the patient data set, which was significantly better than the other comparison methods (permutation P-value=0.001) (Table 1, Figure 2). Exomiser and CADD score-based analysis of the data from the unsolved cases (N=33) did not identify any additional diagnoses.

Comparison of the performance of four variant prioritization tools using data from 47 patients diagnosed through singleton WES.
Clinicians included 40 out of the 49 WES diagnoses in their list of differential diagnoses (82%). The lists generated by PhenoTips and Phenomizer contained 14 (28%) and 18 (37%) of these diagnoses respectively.
Discussion
A key challenge in singleton WES analysis is selection of variants for curation. In most laboratories, this task would be completed by a laboratory scientist. If characteristics of the variant alone guide variant selection, a large number of variants would require curation, increasing the time required for WES data analysis. Even if the scientist utilizes the clinical information provided at the time of test request, this is often brief and may not provide sufficient detail to assist with variant selection. This study demonstrates the continuing value of clinician-generated candidate gene lists in increasing the efficiency of singleton WES data analysis. Using clinical information resulted in ranking causative variants an average of 8.2 positions higher than prioritization based on variant properties alone, reducing the curation burden for laboratory scientists. This clinically driven prioritization framework significantly outperformed purely computational tools, placing a greater proportion of causative variants top or in the top 5 (permutation P-value=0.001).
Exomiser performed the best of the computational tools, slightly outperforming CADD and Condel scores in placing a higher proportion of causative variants top (37% as top hit vs 27% as ranked by CADD score, and 9% as ranked by Condel score (missense variants only)), highlighting the value of incorporating phenotype matching in variant prioritization. The performance of Exomiser in our hands is below that previously published17 where artificially seeded causative variants were ranked top for 96–97% of 10 000 simulated exomes, and 54% of 11 causative variants were ranked as top using data from 9 previously diagnosed patients from the National Institutes of Health (NIH) Undiagnosed Diseases Program (UDP) when family member data were used and 45% as top hit when family data were not used. These differences in performance may in part be due to the nature of our infant cohort, which included a large number of early and relatively undifferentiated clinical presentations. This is in contrast to the 9 families from the NIH Undiagnosed Diseases Program, which are presumably at the end of the diagnostic trajectory. Conversely, although our strategy is effective in patients in whom a differential diagnosis list can be generated by clinicians, almost by definition it is not applicable in the setting of an undiagnosed diseases programme by virtue of patients only being included when clinician input has been exhausted.
Despite the strong performance of the gene list approach, restricting WES data analysis to candidate gene lists would result in a significant proportion of diagnoses being missed. In this cohort, 20% of WES diagnoses were due to variants in genes outside of the clinician-generated candidate gene lists. Four such cases involved genes that were only associated with disease in the medical literature after the original gene list was compiled, and the other five cases involved atypical, complex and very rare diagnoses that were not considered in the clinical differential diagnosis but in retrospect reconciled the patient’s features.
The generation of candidate gene lists for each patient undergoing WES by clinicians is a labour-intensive task, and adds considerable time per patient to the typical clinical workload. This has decreased over time, as we have developed standard gene lists for common presentations such as microcephaly and with the emergence of gene list tools such as the Genomics England gene panel app (https://bioinfo.extge.co.uk/crowdsourcing/PanelApp/). However, in our experience, a small proportion of patients (predominantly those with dysmorphic syndromes or complex phenotypes) still benefit from customized lists. Multidisciplinary peer review of cases at dedicated meetings is utilized at a number of centres to facilitate appropriate patient selection for genomic testing.18 We have utilized this type of structure for candidate gene list refinement as well as patient selection. While this was initially an additional time- and labour-intensive activity, our experience is that it can be successfully incorporated into pre-existing clinical activities that historically served a similar purpose (dysmorphology case review meeting).
Given the increase in clinical workload mandated by this approach, the potential to introduce clinician bias into data analysis, and concerns regarding sustainability, scalability and inter-clinician variability, we were keen to evaluate whether computational tools can replace and/or outperform clinicians in generating lists of differential diagnoses and/or candidate genes. Candidate gene lists based on patient phenotype have been reported as having been used in other studies. These have been generated by laboratories using phenotypic information supplied on requisition forms by searching databases such as the Human Gene Mutation Database and Online Mendelian Inheritance in Man;8 or in the case of the study by Willig et al, by using Phenomizer,12, 19 resulting in candidate gene lists of over 200 genes. The candidate gene lists for the patients included in this study varied in length from 1 to 161 genes, but most were relatively short (median of 23), reflecting firstly the presence of multiple physical and other abnormalities in this patient group, which assist in narrowing the differential diagnosis; and secondly, critical appraisal by experienced clinicians familiar with the patient. Furthermore, we found that while clinicians included 40 out of the 49 WES diagnoses in their a priori list of differential diagnoses (82%), software tools such as PhenoTips and Phenomizer generated lists that contained 14 (28%) and 18 (37%) of these diagnoses respectively. It is likely this difference in performance is due to factors such as clinician experience, skills such as ‘gestalt’ recognition, and the ability of clinicians to take into account additional factors such as age at presentation, family history and relevant negative findings in deriving differential diagnoses.
Genomic testing places new demands on traditional models for the funding and delivery of laboratory and clinical genetics services. Patient care and laboratory workflows are likely to benefit from the delivery of genomic services by integrated clinical-laboratory centres. To effectively engage in this partnership, clinical geneticists need to develop a deep understanding of genomic data interpretation, and shift their skill set towards interpreting phenotype data in the genomic context. While some of the additional time commitment required may be offset by a reduction in the number of patients undergoing lengthy diagnostic odysseys, genomic testing is also likely to increase the overall number of patients referred to genetic services. The investment required to develop and expand the genomic clinical workforce is also likely to be substantial. It will be important to account for these increased time and workforce demands in funding models, which typically focus on test reimbursement to laboratories.
Although this study represents a comprehensive evaluation of purely computational and combined clinical-computational variant prioritization approaches using real patient data, it is still limited by its small size and does not include evaluation of any proprietary variant prioritization tools. The performance of the clinically-driven framework described is clinician-dependent which may affect reproducibility, and the utility of the gene list approach is likely to be limited with non-specific patient phenotypes such as non-syndromic intellectual disability. We have also not compared the performance of alternative variant ranking tools in combination with clinician-driven gene lists. It is possible that further improvements in overall ranking could be achieved by replacing the VPI with specialized ranking tools such as Exomiser. Incorporating variant frequency data from large data sets such as from the Exome Aggregation Consortium (ExAC)20 can be expected to improve the ability of computational tools such as the VPI to highlight potential de novo variants from singleton WES data particularly for rare paediatric disease diagnosis. We anticipate that ongoing work in this area will see further improvements in variant ranking algorithms and robust clinical evaluation of these methods will also be an important ongoing research activity.
The recently published recommendations regarding the integration of genomics into clinical practice highlight the importance of structured phenotyping and phenotype-driven gene lists in facilitating data interpretation.21 The optimum timing and mode of incorporating phenotype information and clinical expertise into genomic data analysis remains to be determined. Reassessment of WES data by a clinical geneticist familiar with the patient led to a change in diagnosis in 21 of 155 (14%) patients seen in an Exome Clinic, with an overall increase in diagnostic yield of 7%.22 Our study provides quantitative evidence for the benefits of utilizing the clinician’s direct and detailed knowledge of the patient in integrating phenotype information at the start of the laboratory genomic analysis process. While computational tools are important adjuncts in WES data analysis, and their performance will continue to improve, close clinical-laboratory interaction remains a key element in the selection of variants for curation and the successful implementation of clinical genomics.
References
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J : A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014; 46: 310–315.
Gonzalez-Perez A, Lopez-Bigas N : Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum genet 2011; 88: 440–449.
Robinson PN, Kohler S, Oellrich A et al: Improved exome prioritization of disease genes through cross-species phenotype comparison. Genome Res 2014; 24: 340–348.
Sifrim A, Popovic D, Tranchevent LC et al: eXtasy: variant prioritization by genomic data fusion. Nat Methods 2013; 10: 1083–1084.
Singleton MV, Guthery SL, Voelkerding KV et al: Phevor combines multiple biomedical ontologies for accurate identification of disease-causing alleles in single individuals and small nuclear families. Am J Hum Genet 2014; 94: 599–610.
Javed A, Agrawal S, Ng PC : Phen-Gen: combining phenotype and genotype to analyze rare disorders. Nat Methods 2014; 11: 935–937.
Smedley D, Robinson PN : Phenotype-driven strategies for exome prioritization of human Mendelian disease genes. Genome Med 2015; 7: 81.
Lee H, Deignan JL, Dorrani N et al: Clinical exome sequencing for genetic identification of rare Mendelian disorders. Jama 2014; 312: 1880–1887.
Farwell KD, Shahmirzadi L, El-Khechen D et al: Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med 2015; 17: 578–586.
Sadedin SP, Dashnow H, James PA et al: Cpipe: a shared variant detection pipeline designed for diagnostic settings. Genome Med 2015; 7: 68.
Girdea M, Dumitriu S, Fiume M et al: PhenoTips: patient phenotyping software for clinical and research use. Hum Mutat 2013; 34: 1057–1065.
Kohler S, Schulz MH, Krawitz P et al: Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet 2009; 85: 457–464.
Stark Z, Tan TY, Chong B et al: A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med 2016; 18: 1090–1096.
Wang K, Li M, Hakonarson H : ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010; 38: e164.
Fokkema IF, Taschner PE, Schaafsma GC, Celli J, Laros JF, den Dunnen JT : LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 2011; 32: 557–563.
Richards CS, Bale S, Bellissimo DB et al: ACMG recommendations for standards for interpretation and reporting of sequence variations: Revisions 2007. Genet Med 2008; 10: 294–300.
Bone WP, Washington NL, Buske OJ et al: Computational evaluation of exome sequence data using human and model organism phenotypes improves diagnostic efficiency. Genet Med 2015; 18: 608–617.
Ormondroyd E, Mackley MP, Blair E et al: Insights from early experience of a Rare Disease Genomic Medicine Multidisciplinary Team: a qualitative study. Eur J Hum Genet 2017; 25: 680–686.
Willig LK, Petrikin JE, Smith LD et al: Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med 2015; 3: 377–387.
Lek M, Karczewski KJ, Minikel EV et al: Analysis of protein-coding genetic variation in 60706 humans. Nature 2016; 536: 285–291.
Bowdin S, Gilbert A, Bedoukian E et al: Recommendations for the integration of genomics into clinical practice. Genet Med 2016; 18: 1075–1084.
Baldridge D, Heeley J, Vineyard M et al: The Exome Clinic and the role of medical genetics expertise in the interpretation of exome sequencing results. Genet Med 2017; e-pub ahead of print 2 March 2017; doi:10.1038/gim.2016.224.
Acknowledgements
We would like to acknowledge the Melbourne Genomics Health Alliance Steering Group, Genomics and Bioinformatics Advisory Group and the Clinical Interpretation Reporting Group for establishing the systems and standards applied in this study. The study was funded by the founding organizations of the Melbourne Genomics Health Alliance (Royal Melbourne Hospital, Royal Children’s Hospital, University of Melbourne, Walter and Eliza Hall Institute, Murdoch Childrens Research Institute, Australian Genome Research Facility and CSIRO) and the State Government of Victoria (Department of Health and Human Services). The involvement of AGRF was supported by sponsorship from Bioplatforms Australia and the NCRIS program.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Stark, Z., Dashnow, H., Lunke, S. et al. A clinically driven variant prioritization framework outperforms purely computational approaches for the diagnostic analysis of singleton WES data. Eur J Hum Genet 25, 1268–1272 (2017). https://doi.org/10.1038/ejhg.2017.123
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.123
This article is cited by
-
Evaluation and pilot testing of a multidisciplinary model of care to mainstream genomic testing for paediatric inborn errors of immunity
European Journal of Human Genetics (2023)
-
Best practices for the interpretation and reporting of clinical whole genome sequencing
npj Genomic Medicine (2022)
-
Rapid and accurate interpretation of clinical exomes using Phenoxome: a computational phenotype-driven approach
European Journal of Human Genetics (2019)
-
Early diagnosis of Pearson syndrome in neonatal intensive care following rapid mitochondrial genome sequencing in tandem with exome sequencing
European Journal of Human Genetics (2019)
