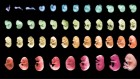- TECHNOLOGY FEATURE
Revealing chromosome contours, one dot at a time

3D reconstruction of a nucleus. Each colour represents a different chromosome, and each dot is a gene location. Credit: Y. Takei et al./Science
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 602, 713-715 (2022)
doi: https://doi.org/10.1038/d41586-022-00496-7
References
Flavahan, W. A. et al. Nature 529, 110–114 (2016).
Zazhytska, M. et al. Preprint at bioRxiv https://doi.org/10.1101/2021.02.09.430314 (2021).
Lubeck, E., Coskun, A. F., Zhiyentayev, T., Ahmad, M. & Cai, L. Nature Methods 11, 360–361 (2014).
Takei, Y. et al. Science 374, 586–594 (2021).
Nguyen, H. Q. et al. Nature Methods 17, 822–832 (2020).
Quinodoz, S. A. et al. Cell 174, 744–757 (2018).
Payne, A. C. et al. Science 371, eaay3446 (2020).
Su, J.-H., Zheng, P., Kinrot, S. S., Bintu, B. & Zhuang, X. Cell 182, 1641–1659 (2020).
 Single-cell analysis enters the multiomics age
Single-cell analysis enters the multiomics age
 Starfish enterprise: finding RNA patterns in single cells
Starfish enterprise: finding RNA patterns in single cells
 Method of the Year: spatially resolved transcriptomics
Method of the Year: spatially resolved transcriptomics
 Single-cell proteomics takes centre stage
Single-cell proteomics takes centre stage
 NatureTech hub
NatureTech hub