- NEWS AND VIEWS
Small molecule combats cancer-causing KRAS protein at last
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 575, 294-295 (2019)
doi: https://doi.org/10.1038/d41586-019-03242-8
References
Sanchez-Vega, F. et al. Cell 173, 321–337 (2018).
Canon, J. et al. Nature https://doi.org/10.1038/s41586-019-1694-1 (2019).
Lu, S., Jang, H., Nussinov, R. & Zhang, J. Sci. Rep. 6, 21949 (2016).
AACR Project GENIE Consortium. Cancer Discov. 7, 818–831 (2017).
Herbst, R. S., Morgensztern, D. & Boshoff, C. Nature 553, 446–454 (2018).
Ostrem, J. M., Peters, U., Sos, M. L., Wells, J. A. & Shokat, K. M. Nature 503, 548–551 (2013).
Janes, M. R. et al. Cell 172, 578–589 (2018).
Patricelli, M. P. et al. Cancer Discov. 6, 316–329 (2016).
Fakih, M., O’Neil, B. & Price, T. J. J. Clin. Oncol. 37 (suppl.), 3003 (2019).
Govindan, R. et al. Ann. Oncol. 30 (Suppl. 5), mdz244.008 (2019).
Hallin, J. et al. Cancer Discov. https://doi.org/10.1158/2159-8290.CD-19-1167 (2019).
Competing Interests
R.S.H. has received honoraria from the following:
Consulting: Abbvie Pharmaceuticals, ARMO Biosciences, AstraZeneca, Biodesix, Bolt Biotherapeutics, Bristol-Myers Squibb, Eli Lilly and Company, EMD Serrano, Genentech/Roche, Genmab, Halozyme, Heat Biologics, IMAB Biopharma, Immunocore, Infinity Pharmaceuticals, Loxo Oncology, Merck and Company, Midas Health Analytics, Nektar, Neon Therapeutics, NextCure, Novartis, Pfizer, Sanofi, Seattle Genetics, Shire PLC, Spectrum Pharmaceuticals, Symphogen, Takeda, Tesaro, Tocagen.
Research support: AstraZeneca, Eli Lilly and Company, Merck and Company.
He is a member of the board of directors (non-executive/ independent) for Junshi Pharmaceuticals.
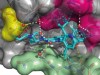 Read the paper: The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity
Read the paper: The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity
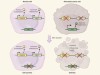 Double trouble for tumours
Double trouble for tumours
 Many mutations in one clinical-trial basket
Many mutations in one clinical-trial basket