- OUTLOOK
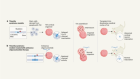
Patisiran’s path to approval as an RNA therapy

Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 574, S7 (2019)
doi: https://doi.org/10.1038/d41586-019-03070-w
This article is part of Nature Outlook: RNA therapy, an editorially independent supplement produced with the financial support of third parties. About this content.
 RNA therapies explained
RNA therapies explained
 Pharma’s roller-coaster relationship with RNA therapies
Pharma’s roller-coaster relationship with RNA therapies
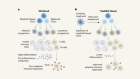
 The challenge of delivering RNA-interference therapeutics to their target cells
The challenge of delivering RNA-interference therapeutics to their target cells
 Unlocking the potential of vaccines built on messenger RNA
Unlocking the potential of vaccines built on messenger RNA
 How RNA therapies could be used to tackle the world’s biggest killer
How RNA therapies could be used to tackle the world’s biggest killer
 It’s time for scientists to shout about RNA therapies
It’s time for scientists to shout about RNA therapies
 Why rare genetic diseases are a logical focus for RNA therapies
Why rare genetic diseases are a logical focus for RNA therapies