- NEWS AND VIEWS
Fluorinated compounds present opportunities for drug discovery
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 573, 37-38 (2019)
doi: https://doi.org/10.1038/d41586-019-02611-7
References
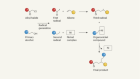
Scattolin, T., Bouayad-Gervais, S. & Schoenebeck, F. Nature 573, 102–107 (2019).
Purser, S., Moore, P. R., Swallow, S. & Gouverneur, V. Chem. Soc. Rev. 37, 320–330 (2008).
Tyrra, W. J. Fluor. Chem. 109, 189–194 (2001).
Scattolin, T., Deckers, K. & Schoenebeck, F. Angew. Chem. Int. Edn 56, 221–224 (2017).
Eckert, H. & Forster, B. Angew. Chem. Int. Edn 26, 894–895 (1987).
Yamaguchi, K. et al. J. Am. Chem. Soc. 113, 5474–5475 (1991).
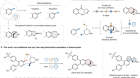
 Read the paper: Straightforward access to N-trifluoromethyl amides, carbamates, thiocarbamates and ureas
Read the paper: Straightforward access to N-trifluoromethyl amides, carbamates, thiocarbamates and ureas
 A radical approach to diversity
A radical approach to diversity
 A cure for catalyst poisoning
A cure for catalyst poisoning