Libraries of virtual compounds could help uncover new drugs.Credit: Laurence Dutton/Getty
Drug discovery is a notoriously tough process. Pharmaceutical companies tend to prize efficiency, so many potential lead compounds are merely iterations of what the companies already have, dictated by what they already know, and rely on already exploited molecular scaffolds (the core structure of a molecule).
The need to diversify molecular scaffolds to improve the chances of success in drug discovery has been referred to as escaping from ‘flatland’ — the reliance on synthetic methods that build flat molecules. Another way to investigate the unexplored potential in the molecular universe is to find a way to reveal what is hidden in the shadows. Some estimates say that there are at least 1060 different drug-like molecules: a novemdecillion of possibilities. How, then, to open up more of this dark chemical space?
Read the paper: Ultra-large library docking for discovering new chemotypes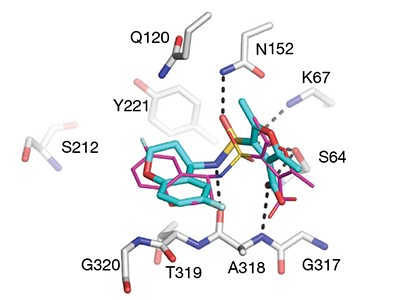
A paper published this week demonstrates the power of ultra-large virtual libraries in helping researchers to look into the unknown (J. Lyu et al. Nature https://doi.org/10.1038/s41586-019-0917-9; 2019). In it, the authors built a virtual library of around 350 million drug-like molecules. They used this to simulate the ways that these molecules could interact with two therapeutically relevant proteins — AmpC β-lactamase, a target for antibiotics, and the D4 dopamine receptor, linked to several neurological disorders and a member of the pharmacologically important family of G protein-coupled receptors.
After this virtual screening, the team synthesized the top-scoring compounds and tested them against the two targets. One of the compounds turned out to be the most potent inhibitor of AmpC β-lactamase known, and is chemically distinct from all other known inhibitors.
Of the 500 or so molecules the group made that targeted the D4 dopamine receptor, one had an unprecedented ability to stimulate it. This compound’s selectivity over other dopamine receptor types, and its preferential activation of the G protein signalling pathway, are both important properties that might help to minimize unwanted side effects when, and if, it’s used as a drug.
News & Views: Bigger is better in virtual drug screens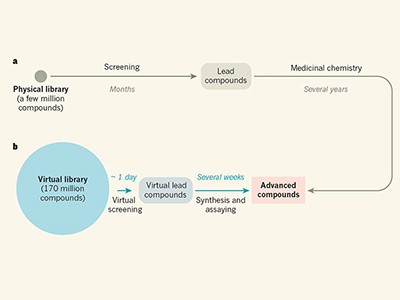
Others have already demonstrated the potential of smaller virtual libraries to aid drug design. But as the accompanying News and Views (D. E. Gloriam Nature https://doi.org/10.1038/d41586-019-00145-6; 2019) shows, the increased library size makes an important difference. The publicly available library (http://zinc15.docking.org) is anticipated to increase to more than a billion molecules within two years.
Going from a promising compound to an approved drug is still a tortuous and uncertain process. But by having access to a greater portion of the chemical universe, the chances of discovering a star should be greater too.
 Read the paper: Ultra-large library docking for discovering new chemotypes
Read the paper: Ultra-large library docking for discovering new chemotypes
 News & Views: Bigger is better in virtual drug screens
News & Views: Bigger is better in virtual drug screens
 How to explore chemical space using algorithms and automation
How to explore chemical space using algorithms and automation
 The drug-maker’s guide to the galaxy
The drug-maker’s guide to the galaxy
 Reaction combination opens up 3D molecular diversity for drug discovery
Reaction combination opens up 3D molecular diversity for drug discovery