Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Rent or buy this article
Prices vary by article type
from$1.95
to$39.95
Prices may be subject to local taxes which are calculated during checkout
Nature 561, 319-320 (2018)
doi: https://doi.org/10.1038/d41586-018-05883-7
Updates & Corrections
-
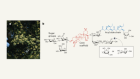
Correction 08 March 2019: In view of the fact that the authors of ‘A homing system targets therapeutic T cells to brain cancer’ (H. Samaha et al. Nature 561, 331–337; 2018) are retracting their report, Michael Platten wishes to retract the News & Views article ‘T cells engineered to home in on brain cancer’ (M. Patten Nature 561, 319–320; 2018), which dealt with this study and was based on the accuracy and reproducibility of their data.
References
Mellman, I., Coukos, G. & Dranoff, G. Nature 480, 480–489 (2011).
Platten, M. & Reardon, D. A. Semin. Neurol. 38, 62–72 (2018).
Thorsson, V. et al. Immunity 48, 812–830 (2018).
Samaha, H. et al. Nature 561, 331–337 (2018).
Engelhardt, B., Vajkoczy, P. & Weller, R. O. Nature Immunol. 18, 123–131 (2017)
Ransohoff, R. M. & Engelhardt, B. Nature Rev. Immunol. 12, 623–635 (2012).
Quail, D. F. & Joyce, J. A. Cancer Cell 13, 326–341 (2017).
Hu, X. et al. Nature Commun. 7, 13095 (2016).
June, C. H. & Sadelain, M. N. Engl. J. Med. 379, 64–73 (2018).
Weller, M. et al. Nature Rev. Neurol. 13, 363–374 (2017).
 Keeping it real to kill glioblastoma
Keeping it real to kill glioblastoma
 Dendritic-cell vaccines on the move
Dendritic-cell vaccines on the move