
Abstract
Huntington’s disease (HD) is a devastating neurodegenerative disorder whose main hallmark is brain atrophy. However, several peripheral organs are considerably affected and their symptoms may, in fact, manifest before those resulting from brain pathology. HD is of genetic origin and caused by a mutation in the huntingtin gene. The mutated protein has detrimental effects on cell survival, but whether the mutation leads to a gain of toxic function or a loss of function of the altered protein is still highly controversial. Most currently used in vitro models have been designed, to a large extent, to investigate the effects of the aggregation process in neuronal-like cells. However, as the pathology involves several other organs, new in vitro models are critically needed to take into account the deleterious effects of mutant huntingtin in peripheral tissues, and thus to identify new targets that could lead to more effective clinical interventions in the early course of the disease. This review aims to present current in vitro models of HD pathology and to discuss the knowledge that has been gained from these studies as well as the new in vitro tools that have been developed, which should reflect the more global view that we now have of the disease.
Similar content being viewed by others


Facts
-
1
The presence of a mutation in the huntingtin (Htt) gene causes alteration in normal cellular functions, through a gain or a loss of function of the protein.
-
2
The mutant huntingtin protein (mHtt) is ubiquitously expressed in the body.
-
3
mHtt aggregates are found in different cell types and can have detrimental effects on these cells, eventually leading to cell death.
-
4
mHtt has been studied in different in vitro models, which have provided significant insights into the mechanisms underlying the toxicity of the protein. However, new models are urgently needed to study the effects of mHtt in non-neuronal cells, which may account for several of the peripheral features now recognized in the disease.
Open Questions
-
1
What are the consequences of the expression of mHtt in other brain cell populations and to what extent it contributes to brain degeneration?
-
2
What is the impact of mHtt expression in different cell populations of the body?
-
3
What are the interactions of mHtt with proteins associated with other diseases?
Huntington’s disease (HD) is a devastating neurodegenerative disorder characterized clinically by a triad of movement disorders, cognitive dysfunction and psychiatric problems.1 The symptoms are generally triggered in mid-life by an autosomal dominant mutation in the huntingtin (Htt) gene localized on the short arm of chromosome 4. The abnormal gene presents an extended number of CAG repeats which are translated into a polyglutamine (polyQ) stretch near the N-terminus of the protein.2 The mutation leads to the formation of intracellular aggregates that are ubiquitously found in the central nervous system and peripheral tissues. The massive loss of GABAergic medium spiny neurons is the pathological feature that has been predominantly studied in HD. However, several other brain structures and neuronal cell populations are also targeted by the pathology often from disease onset, all of which are associated with these protein aggregates. Importantly, a number of peripheral problems are now recognized to be associated with the disease.2, 3
To this day, the focus of investigation of the role of mutant huntingtin (mHtt) to HD pathology has largely been on neuronal elements. Considering the fact that the role of mHtt in non-neuronal tissues has received less attention, we have deemed relevant to review the current knowledge on the role of mHtt protein in a more global perspective, with the expectation to gain a better understanding of the disease. More specifically, we will discuss the development of new cellular tools to characterize the functional and molecular mechanisms underlying mHtt toxicity, with a particular emphasis on its role in cell types other than neurons. We have reviewed the literature from this new angle with the hope of improving the current in vitro models, further allowing for better drug screens.
The Htt Protein
Normal Htt
Despite a sizable number of studies, the role of the Htt protein remains elusive and has led to intensive debates. Htt is a soluble protein that is ubiquitously expressed, but is present in higher concentrations particularly in the brain.4 This cytoplasmic protein is associated with several organelles, microtubules and vesicular membranes, pointing to a role of the protein in intracellular trafficking,5 exocytosis and endocytosis.6 Htt is also associated with proteins involved in synaptic functions.7 It presents antiapoptotic properties and plays a critical role in embryonic development.8 If overexpressed in various systems (cultured striatal cells, primary cultures from HD mouse models9 or in vivo), normal Htt protects against apoptosis and excitotoxicity.9, 10
mHtt protein
Expanded CAG repeats in the exon 1 of the gene translate into longer polyQ stretches. HD manifests when >40 CAG repeats are present,11 the onset of which has, in fact, been reported to inversely correlate with the number of repeats.12 The polyQ stretch is released following proteolysis and is more toxic when cleaved than when bound to the whole protein. This has been demonstrated in vivo in the first developed genetic model of HD, the R6/2 mouse, generated by the expression of 150 CAG repeats of exon 1 of the human mHtt gene.13 The released polyQ stretches trigger the formation of aggregates, as observed in brain sections immunostained for the polyQ domain of the protein.14 The presence of an expanded polyQ stretch has further been speculated to alter the Htt structure, thereby leading to its cytotoxicity. Although this is the favored hypothesis, others have proposed that longer polyQ stretches are more prone to form insoluble aggregates.15 PolyQ stretches may be found in the cytosol as soluble proteins that can further form oligomers and fibrillary structures.16 When a certain threshold in intracellular polyQ concentration is reached, these homopolymers form insoluble aggregates.17 However, whether the aggregates are cytotoxic per se is not clear. In other disease contexts, such as in Alzheimer’s disease, small oligomers or short fibrillar species are more toxic than amyloid plaques.18, 19, 20 The cytotoxic effect of mHtt takes various forms that affect several cellular pathways and functions. The pleiotropic effects of mHtt cytotoxicity have been reported in a number of HD models (Saccharomyces cerevisiae, Drosophila melanogaster, Caenorhabditis elegans21) and in various cell populations using gene arrays (for review, see Sassone et al.22). The exact mechanisms involved have not yet been unraveled, but it is now well known that post-translational modifications can alter the function of the protein and that the mutated protein interferes with the regulation of gene transcription.22
The Controversy about the Gain or Loss of Function of mHtt
Gain of function
The fact that HD is an autosomal dominant disorder argues in favor of a gain of function of the protein, where mHtt is endowed with a novel, albeit deleterious function that is important to HD pathogenesis. In this view, the mutated form of Htt acquires the ability to interact with several proteins23 (e.g., huntingtin-associated protein (HAP1),24 huntingtin-interacting protein (HIP1),25 G-protein-coupled receptor kinase-interacting protein (GIT1),26 postsynaptic density protein (PSD95),27 etc.). Both the role of the interacting partners and the mHtt function can thus be modified. It has been shown that longer polyQ tracts can promote their self-cleavage. In vivo studies have demonstrated that loss of a single Htt allele is not predictive of the appearance of the pathological phenotype.28 The presence of the normal allele in pathological conditions does not improve the phenotype, that is, homozygous and heterozygous HD patients are indistinguishable.29
Loss of function
The presence of mHtt can also lead to a loss of function followed by impairments of mitochondrial integrity and vesicular trafficking. For instance, mitochondrial motility is altered and the organelle is eventually immobilized in neurons, as seen in HD models.30 Another important event that is induced by mHtt is the impairment of synthesis and release of brain-derived neurotrophic factor (BDNF), which may significantly contribute to poor neuronal survival.8, 31 The function of normal Htt can further be challenged if sequestered by the mutant protein.29 Considering its antiapoptotic properties and its role in embryonic development, expression of the normal protein is essential for proper function of the organism. In fact, the conditional knockout of the normal gene triggers neuronal degeneration in the postnatal brain.32 Interestingly, absence of the expanded CAG repeats leads to enhanced levels of autophagy in vitro and through this the removal of mHtt neuropil aggregates.33 Similarly, the deletion of the polyQ stretch in HD mice provokes a reduction in aggregate formation, an improvement in motor phenotype and longevity.34 All of this suggests that the presence of the extended polyQ repeat may inhibit the normal function of Htt in autophagy.
Thus, there is evidence for both a gain of toxic function and a loss of normal function of Htt. In HD pathogenesis, the loss of normal Htt function and the response activated by the mutated protein may act in concert and contribute to the development of the disease. There may conceivably be a cross-talk between the two Htt species, which would result in a negative-dominant effect of the altered protein.8, 35, 36 The idea that Htt may undergo both a loss and a gain of function is progressively becoming a prevalent view in the literature.8
In Vitro Models
Cell lines and primary cultures
In vitro models are powerful tools to study and understand the hallmarks of any pathology and to dissect the mechanisms through which various neurodegenerative processes take place. They are also invaluable for integrating observations collected in vivo. Importantly, the use of different mammalian cell types in the mitotic phase enables to investigate the implications of the pathology in a variety of tissues, including peripheral organs in which the expression and impact of HD pathology is significant22 and less understood. Several cell lines have thus been used to model the pathological features of HD, such as the non-neuronal human HeLa cells, the human embryonic (HEK293T) and monkey kidney fibroblast cell lines (COS-7) as well as the Neuro2a (N2a) neuroblastoma (mouse) and neuron-like PC12 (rat) cells, a summary of which is presented in Table 1.
Inducible systems
Different inducible cell models of HD have also been developed to study the expression of the mutated gene and the formation of aggregates in a more controlled manner, including the ability to prevent the otherwise inexorable cell death that results from mHtt toxicity. With such regulatable models, the expression and study of the gene is time- and inducer-dependent. Several cell lines have been used for this purpose, mostly with neuronal phenotypes to more faithfully reproduce the different features of HD pathology. These various models are of key importance for the improvement and development of new cell lines and are presented in Table 2.
What Have We Learned from In Vitro Models?
mHtt proteolysis
Proteolysis is one of the major events that is triggered in cells coping with the misfolding and aggregation of proteins. PolyQ stretches derived from both the normal and mutated protein may undergo degradation. Although initiation of the aggregation process requires mHtt protein, aggregates can in turn recruit polypeptides with the wild-type polyQ stretch.37 The mHtt constantly undergoes cleavage by calpains, caspases38 and metalloproteases, which release smaller N-terminal fragments that can subsequently access the nucleus.39 Indeed, cleavage inhibition results in reduced mHtt toxicity.39 Interestingly, in both in vitro and in vivo models, aggregates are formed more rapidly in cells expressing the truncated form of the mHtt, which carries only the first exon of the gene.13 Cleavage is also influenced by the number of CAG repeats40 as well as the cell type used. When in vitro expression is limited to the short segment of the protein, the pathological features such as protein aggregation and cell death occur more rapidly, suggesting that the N-terminal portion of mHtt is more toxic than the full-length protein.41 These results have also been validated in vivo in the R6/2 mouse, which expresses the truncated human Htt gene. The human HEK293T cells express aggregates when they are transfected with the shorter form of the gene.42, 43 Similarly, it has been observed that full or short forms of the mHtt have different effects in transduced primary cultures derived from mouse E17 embryonic striatal cells.44 Early post-transduction, most cells expressing the truncated form of the mHtt gene develop aggregates. All cells eventually contain inclusions. Conversely, cells harboring full-length mHtt express few aggregates. Notably, the aggregates derived from the short form of the Htt gene are detected in both the cytosol and nucleus early after transfection. At later stages, aggregates localize exclusively in the nucleus. Cells expressing the full-length gene are characterized by cytosolic inclusions, in contrast with the nuclear localization that is observed in vivo, although this may be due to a shorter follow-up of the aggregation process in cultured cells.44 Importantly, aggregate formation leads to cell death, thus mimicking the pathological features of the disease, as shown both in neuronal primary cultures45 and differentiated neuronal cell lines46 such as PC12 cells. However, the expression level of the mutated protein in non-neuronal elements such as in HEK293T cells is not correlated with cytotoxic effects. This may be due to the mitotic status of the cells, which entails a dilution of protein content in daughter cells.
The discovery of mHtt cleavage has been an important milestone in our understanding of the pathological events underlying HD. It has also provided a tool for studying the protein and the aggregation process in vitro over brief periods of time corresponding to a few cell cycles, that is, the normal lifespan of a cultured cell. However, it remains difficult to determine the balance between the cleaved and uncleaved forms of the protein, as well as between the soluble and aggregated forms, which are factors that may contribute to a slower appearance of some of the characteristics of the pathology in vivo. Indeed, shorter fragments aggregate faster and have higher rates of nuclear translocation. This could be due to the smaller dimensions of the fragments that may more easily be translocated through the nuclear pores. The variable lengths of the mHtt polypeptide species also influence the cellular localization of the protein in vitro and thus its cytotoxic effects.40 Furthermore, the C-terminal domain of the protein has been found within inclusions in vivo, but epitopes of this protein fragment can be hidden by other portions of the protein. Thus, it cannot be excluded as a potential event involved in the aggregation process.
Aggregate formation
Aggregation is a dynamic process.47, 48 Unlike in normal cells, where the Htt protein is found primarily in the cytoplasm, aggregates are found in both the nucleus and cytoplasm in pathological conditions. However, the consequences of intracellular mHtt accumulation are still highly debated.49, 50 Purified mHtt fragments reveal that shorter polyQ stretches of the protein tend to adopt a β-structure (β-strand/β-turn), whereas longer polyQ chains form more globular structures, similar to those yielded by a fibrillization process. Globular intermediates can further form protofibrils, which ultimately lead to mature fibers. Conformational changes are important determinants of the protein toxicity.51 Observations collected in a new model of mHtt aggregation, developed in transiently transfected COS-7, have led to the idea of a dynamic four-step process to explain protein aggregation.52 The first phase is the accumulation phase, during which only soluble misfolded monomers are present at low concentrations. In the second phase, small oligomers are formed, which remain in equilibrium with monomers. During the third phase, nucleation is triggered at one or more cytoplasmic sites (i.e., nucleation centers), ultimately leading to the formation of larger inclusions, the fourth phase of aggregation.52 Normal Htt can also contribute to the formation of aggregates,53 providing some explanation as to why the normal protein does not buffer the mutated protein and thus cannot have a protective action. In this context, inducible systems may be useful tools to unravel the relationship between dose-dependent expression of mHtt and the formation of aggregates.
However, not all current evidence supports the idea that aggregates are toxic. Arrasate et al.54 demonstrated that neurons die according to the amount of polyQ protein present within neuronal cells in a dose-dependent but time-independent manner.54 Notably, the authors have demonstrated that dying cells do not always contain aggregates, and that, in fact, the presence of aggregates increases the cell’s lifespan. Preventing aggregate formation results in a higher percentage of cell death.55, 56, 57 These observations have led to the hypothesis that aggregate formation is necessary to sequester pathogenic forms of the protein.
Investigation of the subcellular localization of mHtt in various cell types
The most common in vitro tools used to study HD pathology are neuronal cell lines. The use of cells that can differentiate and be maintained in a postmitotic phase enables comparison between dividing and non-dividing cells of the same clonal origin. For example, Wyttenbach et al.46 developed a tetracycline (Tet)-ON-inducible PC12 cell line that, in the absence of the inducer doxycycline, expresses the construct at low levels. In this system, undifferentiated cells show aggregates in the cytosol and nucleus while, upon differentiation into a neuronal phenotype, they present only rare occurrences of nuclear inclusions in cell soma or neurites. The expression of aggregates within neurites can further inhibit outgrowth, which then leads to death.46 These observations have been corroborated in in vivo models where aggregate accumulation is observed in dystrophic neurites.58
The N2a cell line has been very useful to investigate the increased susceptibility of differentiated cells to death. Indeed, the differentiated neurons are seemingly more vulnerable in that respect to the presence of aggregates.50, 59 As described in Pennuto et al.,60 mHtt is also subject to post-translational modifications that can influence the localization and therefore the toxicity of the protein. More specifically, nuclear localization is influenced by phosphorylation of the Ser-13 and Ser-16 located in the N-terminal domain.61 By point mutation, Havel et al.62 have demonstrated that Ser-16 is essential for both aggregation and nuclear localization, as phosphorylation reduces the affinity of the protein for nuclear pores, which are involved in the nuclear transport pathway. Importantly, striatal neurons of the full-length knock-in (140 CAG) HD animal model show an increased phosphorylation of S16. A recent study, also performed in striatal cell lines (STHdH), has demonstrated the importance of post-translational modifications in the first 17 N-terminal amino acids of the protein in mHtt localization.
Cells are selectively susceptible to the formation of aggregates, not only because of the expression of the full-length or truncated protein but also because of the site of aggregate compartmentalization within the cells. The contribution of intracellular localization and intramolecular interactions to aggregate toxicity remains an important issue to explore.
Protein–protein interaction
Htt has been shown to interact with proteins involved in various cellular pathways. The capacity of mHtt to interact with numerous partners resides primarily in its unique structure.63 When a mutation occurs and a longer polyQ stretch is present, the protein undergoes conformational changes, culminating in altered or even the loss of protein–protein interactions. The length of the polyQ stretch and its solubility status do not affect protein interactions. However, the presence of aggregates can lead to changes in the distribution and solubility of mHtt-sequestered partners.64 In fact, essential proteins may be recruited within aggregates and thereby become physiologically inactive, or displaced within the cell, thus impacting normal cellular function.65 Moreover, aggregate formation triggers the transient recruitment of chaperones (e.g., heat-shock protein-70 and heat-shock protein-40) and proteasomes, potentially as a cellular reaction to remove aggregates.66, 67, 68, 69, 70 Likewise, polyQ-containing proteins are irreversibly trapped, leading to a loss of function. Physiological partners of Htt such as HIP1 or HAP1 may also be sequestered into aggregates.64
Mounting evidence implicates numerous protein interactions in aggregate formation, a phenomenon also now recognized in different diseases. Importantly, some proteins are consistently identified in aggregates found in various diseases. For instance, α-synuclein (α-syn), a protein widely associated with Lewy bodies found in Parkinson’s disease, has been detected in tauopathies, β-amyloid plaque and neuritic tangles,71, 72, 73 and more recently within mHtt aggregates, as revealed both by in vitro and post-mortem studies.74, 75, 76 The overexpression of the full-length α-syn in PC12 cells promotes and increases mHtt exon1 aggregation. Alone, α-syn does not aggregate and is not found in mHtt aggregates, suggesting that it rather acts as a seed (or nucleating center) for mHtt aggregation.77 Using a fluorescent bimolecular complement assay in human neuroglioma cells, α-syn and mHtt have been shown to interact and co-aggregate in large inclusions. However, the presence of α-syn does not reduce the toxicity of mHtt aggregates.78 In fact, α-syn knockout cells display a decreased number of mHtt aggregates in transfected mouse neurons, likely as a result of increased autophagy.76 The disease-modifying properties of α-syn have been further demonstrated in HD, revealing the contribution of the protein to disease progression. Tau has recently been reported to colocalize in mHtt aggregates.79 However, the exact role of this interaction is still not clear. More recently, an interaction between optineurin and Htt has been observed in post-mortem HD brain tissue.80 Optineurin is associated with some familial forms of amyotrophic lateral sclerosis and it is ubiquitously expressed both in the brain and in the periphery. In HD brains, optineurin is localized within intranuclear, perykarial and cytoplasmic inclusions. Other studies have reported cytoplasmic localization of optineurin with mHtt inclusions, but not in the nucleus. This might be due to differences in the cerebral areas analyzed.81 It is not clear whether optineurin contributes to the formation of aggregates, but its interaction with mHtt may indicate an involvement in the etiology of the disease. The identification and characterization of the potential interactions between mHtt and other proteins is crucial for a better comprehension of disease progression. In addition, it may lead to the discovery of new common pathways among diseases and thereby the development of therapeutic strategies that are profitable to a number of pathologies. Development of new cell lines expressing mHtt, along with other proteins found in disease settings, is critical to this purpose.
The Importance of Htt Expression in Non-Neuronal Cells
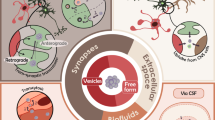
Understanding the pathological role of mHtt is a considerable challenge given its ubiquitous expression in the body. To date, studies have largely excluded the investigation of the potential pathological mechanisms implicated in peripheral abnormalities and have mainly focused on events specific to neuronal cells. However, cellular dysfunction due to mHtt expression and aggregate formation is also prominent in other organs (Table 3 and Figure 1). Skeletal muscle dysfunction, cardiac failure and weight loss are only a few examples of peripheral symptoms found in HD.22 It is thus imperative to develop new cellular models that incorporate these aspects of the pathology. To date, the examples are very few with the restricted use of human embryonic kidney (HEK293T) or monkey kidney fibroblast cell line (COS-7). However, these cells are less prone to apoptotic death as compared to cells in the postmitotic phase. The differential susceptibility of various cell types to mHtt cytotoxicity could be due to the differences in their respective cell cycles. Mature neurons, which no longer replicate, have a greater tendency to display aggregates, although there is no correlation between the level of mHtt expression and the rate of cell death in vivo.82

Schematic representation of various peripheral cell types involved in the pathogenesis of HD and the putative mechanisms that affect normal cell physiology and functions according to the data reported in mice and human tissues, as well as in cell models. Skeletal and heart muscle cells show mHtt aggregate formation that could interfere with mitochondrial functions and disrupt actin and myosin microfilament networks. Endocrine dysfunction may affect pancreatic cells or adipose tissue. Abnormal functions resulting from these changes could impact on disease development and contribute to symptoms such as weight loss. Blood cells, such as monocytes and lymphocytes, display pathological features that are accompanied by an increased inflammatory response and release of proinflammatory cytokines (e.g., interleukin (IL)-6, IL-4, tumor necrosis factor (TNF)-α). Abbreviations: UPS, ubiquitin–proteasome system; ER, endoplasmic reticulum; Q, polyglutamine stretch; Ins, insulin; PGC-1β, peroxisome proliferator-activated receptor γ coactivator 1
Current knowledge on the role of the Htt protein has provided significant insights into the pathology of HD. However, it is crucial to develop new models based on cells from various peripheral tissues where the pathology is also present, but has heretofore been largely ignored. It is fundamental to understand whether the continuous turnover of proliferating cells can dilute the mutated protein in their progeny, thereby reducing the probability of aggregate accumulation. Another aspect of interest is to evaluate whether neuronal and peripheral cells have different requirements for Htt expression. For example, it has been demonstrated that neuronal populations express various levels of Htt protein.83 Interestingly, mHtt mRNA contents vary between cell types.84 Accordingly, this may indicate a different mechanism of regulation of mHtt expression. Thus far, various peripheral tissues from HD patients and mouse models such as muscle, blood and adipose tissue have been studied in vivo.22 The peripheral cell populations investigated display several alterations such as transcriptional dysfunction and mitochondrial deficits, which may represent important new hallmarks and potential biomarkers for HD diagnosis. These observations could also lead to important clues related to the pathology in neuronal phenotypes, thus ultimately contributing to the understanding of the disease progression.
mHtt expression in non-neuronal cells
Studies conducted in different cell populations from either patients or HD mouse models have revealed a number of abnormalities associated with the expression of mHtt in non-neuronal cell types. In the disease itself, early abnormalities are observed in muscular cells of HD patients.85 Myoblasts collected from patients show ultrastructural alterations, as well as greater concentrations of mHtt (in either homozygous or heterozygous individuals) when compared to myoblasts derived from normal individuals. Severe mitochondrial alterations that may impact the energy status are also found in these cells. To account for the latter effects, one should keep in mind that mHtt not only interacts directly with organelles, but can also alter transcription. The lower expression and impaired activity of the transcription factor peroxisome proliferator-activated receptor γ coactivator 1-α (PGC-1α), which is involved in the expression of energy metabolism genes, could also be implicated in the mitochondrial dysfunction observed in these cells.86 Similarly, cultured myoblasts from R6/2 mice show the presence of intranuclear aggregates that mirror the evolution of aggregate formation in in vivo brain and muscle tissues.87 This result suggests a crucial role of mHtt in muscle differentiation, which is further supported by the fact that Htt knockout leads to fetal lethality.88 Functional deficits have also recently been observed in fibroblasts derived from HD patients.89, 90 These cells, as do brain cells, show an increased formation of autophagic vacuoles, suggesting a potential relationship in the development of the disease between the central nervous system and the periphery. However, there are still several phenotypic differences between neuronal and peripheral cells that may be due, for example, to the kinetics of aggregation or the different microenvironments of these cells.90
Interestingly, the number of CAG repeats in immortalized R6/2 fibroblasts is highly unstable and translates into changes that correlate with the number of passages. However, cells isolated from newborn mice show more stable numbers of repeats, which may reflect a somatic instability of the CAG stretch.91 The instability depends on polyQ sequence length, as also demonstrated in lymphoblastoid cell lines derived from HD patients.37 Other organs are affected during the progression of the disease. Livers of HD patients, as well as R6/2 mice, present aggregates, are atrophied and do not function normally.92 Hepatic cells (HepG2) overexpressing mHtt show impairment in metabolic pathways and dysregulation of energy homeostasis, likely as a result of sequestration of the PGC-1α transcription factor by mHtt aggregates.93 The pancreas is also characterized by the presence of mHtt. The pathogenic protein can affect pancreatic vesicular transport as well as insulin production and release.94 A recent study has demonstrated that the loss of HAP1, a protein usually associated with Htt and involved in synaptic and vesicular insulin release in β-cells, could play a major role in the disruption of insulin delivery during HD pathogenesis, both in vivo and in vitro (e.g., NIT-1 cells).95
Normal Htt plays an essential role in blood cell production as well. An in vitro study has highlighted the pivotal function of normal Htt in the differentiation of embryonic stem into hematopoietic cells.96 The absence of the normal protein provokes even more profound abnormalities during hematopoiesis, as witnessed by the limited ability of these cells to expand in culture. The authors suggested that the lack of interactions of mHtt with partner proteins such as HIP1, HIP3 and HAP1, may significantly alter their functions in hematopoietic progenitor cells. In addition, alterations in vesicular transport may contribute to abnormalities observed in the absence of Htt. Thus, these data emphasize the importance of the normal protein in hematopoiesis, further suggesting that the presence of mHtt can drastically alter cell behavior. Furthermore, cultured blood cells from HD patients show several aberrations – including mitochondrial abnormalities97 and increases in caspase activity, especially caspase-3, -8 and -9.98 The survival of these impaired cells correlates with the number of CAG repeats in a time-dependent manner. These observations suggest that the level of cellular dysfunction varies according to genotype (i.e., homozygous versus heterozygous for the mutant allele).99 In fact, homozygous lymphoblasts depict large autophagy vacuoles, a hallmark of the disease, in a manner identical to cells with highly expanded and toxic CAG repeats.97 Interestingly, another upregulated target in HD blood and brain that is affected even at presymptomatic stages is the adenosine receptor A2A, involved in inflammatory responses.100, 101 This gene may represent a novel biomarker and therapeutic target for HD. Lymphoblastoid cell lines from HD patients have also been generated to study cell division, and whether it can affect DNA stability. This could also be detected in patients with the juvenile form of HD. The question now is whether a longer polyQ stretch in glial cells, in the aging process, could lead to more extensive brain damage.37 Additional studies will be required to answer this question, both in vitro, using co-culture approaches, as well as in vivo.
mHtt expression in brain cell populations
mHtt expression in astrocytes
mHtt has been found in glial cell populations such as astrocytes.102 Remarkably, selective overexpression of the mHtt protein in these cells can induce the HD phenotype in mice.103, 104 However, similar overexpression of mHtt by viral infection in vitro does not lead to degeneration of astrocytes, although it does in cultured neurons.102 Astrocytes expressing mHtt display a reactive morphology and exhibit greater cell proliferation and mitochondrial respiratory rates.102 Chou et al.105 also reported the presence of aggregates within the nucleus and cytoplasm of astrocytes, although the size of the aggregates was smaller than those found in neurons. The latter difference may likely be due, again, to the fact that glial cells maintain the ability to divide, which could slow down the aggregation process by diluting mHtt contents.
Another aspect of paramount importance is the effect of mHtt on normal astrocytic function. In HD, the expression of the glutamate transporter 1 (GLT1) and the glutamate–aspartate transporter (GLAST) is impaired in astrocytes, possibly altering their ability to buffer glutamate excitotoxicity and therefore impacting neuronal survival.102, 106 Rapamycin, a drug that stimulates autophagy, can re-establish the level of GLT1 in primary rat astrocyte culture transduced with an adenoviral vector coding for the N-terminal domain of Htt.107 Abnormal transcription and secretion of chemokine (C–C motif) ligand (CCL5/RANTES) has further been reported in astrocytic cells.105 This chemokine is responsible for maintaining neuronal activity and neurite development. In fact, primary cultures of astrocytes derived from R6/2 mice show that not only is CCL5 transcription reduced in HD, but there is also an impairment of the chemokine secretory pathways leading to their intracellular accumulation. The impairment of CCL5 secretion may underlie defective neuron–glia interactions.105 The release of other chemokines or neurotrophic factors by astrocytes could also be defective. It has recently been demonstrated that the expression of mHtt in primary cultures of astrocytes impairs the release of BDNF, disrupting the Golgi complex.108 The altered release of neurotrophic factors, as well as the release of chemokines, can negatively influence neuronal survival. The evaluation of the release of other factors will be an important aspect to consider in future studies, given the implication of astrocytes in the degenerative processes associated with HD.
mHtt expression in immune cells
One aspect that has been scarcely explored with in vitro models is the immunological component of the disease. PET scan as well as post-mortem analyses of HD patients at different stages of the disease have revealed a significant brain immune response.109, 110, 111 The presence of pro-inflammatory cytokines has been detected both in the brain112 and blood, revealing higher levels of interleukin-6 (IL-6) in HD patients.113 The expression of mHtt in microglial cells has been further exemplified by RT-PCR in microglial primary cultures from R6/2 mice,114 although nuclear inclusions were not observed.115 The expression of the mutated protein causes neuronal degeneration, which may trigger a local microglial response. A recent study has uncovered an important relationship between cortico-striatal primary neurons expressing the N-terminal domain of mHtt and wild-type microglial cells. In fact, co-culture systems show that morphologically activated immune cells migrate towards the dystrophic neurites rather than the sites of accumulation of mHtt aggregates. The tendency of microglial cells to gather at neuronal processes was further confirmed in slice cultures. However, there is no evidence of a direct contribution of microglial cells to the HD degenerative process in vitro116 where in fact wild-type microglial cells have only been shown to play a supportive role towards neurons transfected with mHtt. However, mHtt-expressing immune cells may adopt a different phenotype. Microglia density decreases over time in R6/2 mice and microglial cells display characteristic structural abnormalities (such as process fragmentation117), abnormal iron metabolism118 and impairments of the kynurenine pathway.114 Conceivably, mHtt may not only affect the transcription machinery of microglial cells114 but also their capacity to impact the neuronal environment.116 However, the presence of aggregates in microglia themselves has not been reported.102 This observation could be explained by differential levels of ubiquitin–proteasome system (UPS) activity between neurons and glial cells.119 Further exploring UPS function in microglial cells would lead to a better understanding of the role of UPS during disease progression, perhaps further revealing a novel target for therapeutic approaches.
Another important aspect that has not been explored yet in HD is the involvement of caspase activation during the inflammatory response driven by microglia cells. Caspase-3 activation occurs not only during apoptosis but also following an inflammatory response.120 Primary microglial cells from animal models, such as R6/2114 or yeast artificial chromosome (YAC) mice, could represent an efficacious tool to investigate the mechanisms related to the inflammatory response in HD. In addition, the availability of mouse models, which express caspase-3- and -6-resistant mHtt, may shed light onto the mechanisms related to this aspect of the inflammatory response.121
mHtt expression in oligodendrocytes
Impairment in myelin production122 as well as alteration in white matter are characteristics of HD pathology. PGC-1α is an important factor in the transcriptional regulation of cellular metabolism and also plays a pivotal role in early myelination. Importantly, PGC-1α is known to be deregulated by mHtt.123 Primary oligodendrocytes expressing normal or mHtt exhibit a reduced expression of PGC-1α, resulting in lower production of cholesterol and myelin basic protein,124 suggesting the involvement of this protein in the alteration of neuronal myelination in HD. The protective role of PGC-1α overexpression against oxidative stress125, 126 and demyelination124 may offer a therapeutic target for HD.
New avenues for mHtt studies in vitro: inducible pluripotent stem cells
Despite the development of a number of cell lines, the field is still lacking human neuronal cell lines that can be representative of early neuronal differentiation.127 Current in vitro research on mHtt is moving towards the use of induced pluripotent stem cells derived from either HD murine models128, 129 or from patients.127 These cells can be easily collected and somatic cell reprogramming can be performed to achieve patient-specific models of the disease.127 Another report recently demonstrated the feasibility of reprogramming primary fibroblasts from patients. Notably, primary fibroblasts can be easily collected from patients and differentiated into neuronal cells.127, 130 Importantly, mHtt does not impair the process of converting the cells into neuronal progenitors or mature neurons. The cells also show increased and sustainable lysosomal content.127 These new tools will be essential to better understand how the mutated protein affects neuronal cells in a more physiological manner and to further identify the specific factors responsible for the higher susceptibility of striatal cell populations, for example, to degeneration in HD. In addition, they will also be important means for evaluating new treatments that could translate into patient-specific therapies.
Conclusion
The view that mHtt is an important player in the development of HD has been widely challenged in several in vitro and in vivo models. When cleaved, the protein releases polyQ fragments which are highly susceptible to aggregate formation, and of which the exact role is still highly controversial. mHtt has several interacting partners that affect the cell physiology in various and complex ways. The identification of proteins interacting with mHtt will certainly provide new clues to the pathological mechanisms involved in HD.
The development of cellular models of the disease from yeast, cell lines and primary cultures has led to an improved understanding of HD pathology. Taken together, these models have revealed that cultured cells are more susceptible to the presence of the pathological form of the protein when the latter is overexpressed, whereas physiological expression leads to more subtle changes. With a better understanding of the features observed in HD, it is becoming increasingly clear that the study of peripheral cells could provide new insights into the neuronal pathology, as events that take place within peripheral tissues may be reminiscent of the abnormalities observed in the brain. Differences in pathological patterns have already been reported between cell types. These differences may be attributed to their specific microenvironments, to differential protein expression or kinetics in protein aggregation, which differ between cell types.
In vitro studies have also allowed the identification of new elements involved in the toxicity of the Htt protein, which may be used as new targets to develop alternative therapeutic approaches. Finally, adequate in vitro models will be essential in answering crucial questions about the reversibility of HD pathology as well as for testing novel therapeutic compounds.
Abbreviations
- Htt:
-
huntingtin protein
- mHtt:
-
mutant huntingtin protein
- HD:
-
Huntington’s disease
- polyQ:
-
polyglutamine
- HAP1:
-
huntingtin-associated protein 1
- HIP1:
-
huntingtin-interacting protein
- GIT1:
-
G-protein-coupled receptor kinase-interacting protein
- PSD95:
-
postsynaptic density protein 95
- YAC:
-
yeast artificial chromosome
- BDNF:
-
brain-derived neurotrophic factor
- Tet:
-
tetracycline
- α-syn:
-
α-synuclein
- PGC-1α:
-
peroxisome proliferator-activated receptor γ coactivator 1-α
- GLT1:
-
glutamate transporter 1
- CCL5:
-
chemokine (C–C motif) ligand
- IL-6:
-
interleukin-6
- UPS:
-
ubiquitin–proteasome system
References
Rothlind JC, Bylsma FW, Peyser C, Folstein SE, Brandt J . Cognitive and motor correlates of everyday functioning in early Huntington’s disease. J Nerv Ment Dis 1993; 181: 194–199.
Ross CA, Shoulson I . Huntington disease: pathogenesis, biomarkers, and approaches to experimental therapeutics. Parkinsonism Relat Disord 2009; 15 (Suppl 3): S135–S138.
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP . Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol 1985; 44: 559–577.
Cattaneo E, Zuccato C, Tartari M . Normal huntingtin function: an alternative approach to Huntington’s disease. Nat Rev Neurosci 2005; 6: 919–930.
Caviston JP, Holzbaur EL . Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol 2009; 19: 147–155.
Smith R, Brundin P, Li JY . Synaptic dysfunction in Huntington’s disease: a new perspective. Cell Mol Life Sci 2005; 62: 1901–1912.
Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA 2000; 97: 6763–6768.
Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S . Loss of normal huntingtin function: new developments in Huntington’s disease research. Trends Neurosci 2001; 24: 182–188.
Leavitt BR, van Raamsdonk JM, Shehadeh J, Fernandes H, Murphy Z, Graham RK et al. Wild-type huntingtin protects neurons from excitotoxicity. J Neurochem 2006; 96: 1121–1129.
Zhang Y, Li M, Drozda M, Chen M, Ren S, Mejia Sanchez RO et al. Depletion of wild-type huntingtin in mouse models of neurologic diseases. J Neurochem 2003; 87: 101–106.
Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR . The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet 1997; 60: 1202–1210.
Djousse L, Knowlton B, Hayden M, Almqvist EW, Brinkman R, Ross C et al. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am J Med Genet A 2003; 119A: 279–282.
Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996; 87: 493–506.
DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997; 277: 1990–1993.
Wellington CL, Brinkman RR, O’Kusky JR, Hayden MR . Toward understanding the molecular pathology of Huntington’s disease. Brain Pathol 1997; 7: 979–1002.
Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 1997; 90: 549–558.
Huang CC, Faber PW, Persichetti F, Mittal V, Vonsattel JP, MacDonald ME et al. Amyloid formation by mutant huntingtin: threshold, progressivity and recruitment of normal polyglutamine proteins. Somat Cell Mol Genet 1998; 24: 217–233.
Xue WF, Hellewell AL, Gosal WS, Homans SW, Hewitt EW, Radford SE . Fibril fragmentation enhances amyloid cytotoxicity. J Biol Chem 2009; 284: 34272–34282.
Dahlgren KN, Manelli AM, Stine WB, Baker LK, Krafft GA, LaDu MJ . Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem 2002; 277: 32046–32053.
Cenini G, Cecchi C, Pensalfini A, Bonini SA, Ferrari-Toninelli G, Liguri G et al. Generation of reactive oxygen species by beta amyloid fibrils and oligomers involves different intra/extracellular pathways. Amino Acids 2010; 38: 1101–1106.
Sugars KL, Rubinsztein DC . Transcriptional abnormalities in Huntington disease. Trends Genet 2003; 19: 233–238.
Sassone J, Colciago C, Cislaghi G, Silani V, Ciammola A . Huntington’s disease: the current state of research with peripheral tissues. Exp Neurol 2009; 219: 385–397.
Giorgini F, Muchowski PJ . Connecting the dots in Huntington’s disease with protein interaction networks. Genome Biol 2005; 6: 210.
Li SH, Gutekunst CA, Hersch SM, Li XJ . Interaction of huntingtin-associated protein with dynactin P150Glued. J Neurosci 1998; 18: 1261–1269.
Metzler M, Legendre-Guillemin V, Gan L, Chopra V, Kwok A, McPherson PS et al. HIP1 functions in clathrin-mediated endocytosis through binding to clathrin and adaptor protein 2. J Biol Chem 2001; 276: 39271–39276.
Goehler H, Lalowski M, Stelzl U, Waelter S, Stroedicke M, Worm U et al. A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease. Mol Cell 2004; 15: 853–865.
Sun Y, Savanenin A, Reddy PH, Liu YF . Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95. J Biol Chem 2001; 276: 24713–24718.
Housman D . Gain of glutamines, gain of function? Nat Genet 1995; 10: 3–4.
Squitieri F, Almqvist EW, Cannella M, Cislaghi G, Hayden MR . Predictive testing for persons at risk for homozygosity for CAG expansion in the Huntington disease gene. Clin Genet 2003; 64: 524–525.
Trushina E, Dyer RB, Badger JD, Ure D, Eide L, Tran DD et al. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol 2004; 24: 8195–8209.
Zuccato C, Cattaneo E . Role of brain-derived neurotrophic factor in Huntington’s disease. Prog Neurobiol 2007; 81: 294–330.
Dragatsis I, Dietrich P, Zeitlin S . Expression of the Huntingtin-associated protein 1 gene in the developing and adult mouse. Neurosci Lett 2000; 282: 37–40.
Zheng S, Clabough EB, Sarkar S, Futter M, Rubinsztein DC, Zeitlin SO . Deletion of the huntingtin polyglutamine stretch enhances neuronal autophagy and longevity in mice. PLoS Genet 2010; 6: e1000838.
Clabough EB, Zeitlin SO . Deletion of the triplet repeat encoding polyglutamine within the mouse Huntington’s disease gene results in subtle behavioral/motor phenotypes in vivo and elevated levels of ATP with cellular senescence in vitro. Hum Mol Genet 2006; 15: 607–623.
Sipione S, Cattaneo E . Modeling Huntington’s disease in cells, flies, and mice. Mol Neurobiol 2001; 23: 21–51.
Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L et al. Loss of huntingtin-mediated BDNF gene transcription in Huntington’s disease. Science 2001; 293: 493–498.
Cannella M, Maglione V, Martino T, Ragona G, Frati L, Li GM et al. DNA instability in replicating Huntington’s disease lymphoblasts. BMC Med Genet 2009; 10: 11.
Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB et al. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington’s disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci USA 2001; 98: 12784–12789.
Johri A, Beal MF . Hunting-ton for new proteases: MMPs as the new target? Neuron 2010; 67: 171–173.
Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K et al. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet 1998; 18: 150–154.
Weiss A, Klein C, Woodman B, Sathasivam K, Bibel M, Regulier E et al. Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington’s disease. J Neurochem 2008; 104: 846–858.
Hackam AS, Hodgson JG, Singaraja R, Zhang T, Gan L, Gutekunst CA et al. Evidence for both the nucleus and cytoplasm as subcellular sites of pathogenesis in Huntington’s disease in cell culture and in transgenic mice expressing mutant huntingtin. Philos Trans R Soc Lond Ser B 1999; 354: 1047–1055.
Hackam AS, Singaraja R, Zhang T, Gan L, Hayden MR . In vitro evidence for both the nucleus and cytoplasm as subcellular sites of pathogenesis in Huntington’s disease. Hum Mol Genet 1999; 8: 25–33.
Huang B, Schiefer J, Sass C, Kosinski CM, Kochanek S . Inducing huntingtin inclusion formation in primary neuronal cell culture and in vivo by high-capacity adenoviral vectors expressing truncated and full-length huntingtin with polyglutamine expansion. J Gene Med 2008; 10: 269–279.
Zala D, Benchoua A, Brouillet E, Perrin V, Gaillard MC, Zurn AD et al. Progressive and selective striatal degeneration in primary neuronal cultures using lentiviral vector coding for a mutant huntingtin fragment. Neurobiol Dis 2005; 20: 785–798.
Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J et al. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington’s disease. Hum Mol Genet 2001; 10: 1829–1845.
Bates G . Huntingtin aggregation and toxicity in Huntington’s disease. Lancet 2003; 361: 1642–1644.
Hands SL, Wyttenbach A . Neurotoxic protein oligomerisation associated with polyglutamine diseases. Acta Neuropathol 2010; 120: 419–437.
Kaytor MD, Wilkinson KD, Warren ST . Modulating huntingtin half-life alters polyglutamine-dependent aggregate formation and cell toxicity. J Neurochem 2004; 89: 962–973.
Poirier MA, Jiang H, Ross CA . A structure-based analysis of huntingtin mutant polyglutamine aggregation and toxicity: evidence for a compact beta-sheet structure. Hum Mol Genet 2005; 14: 765–774.
Poirier MA, Li H, Macosko J, Cai S, Amzel M, Ross CA . Huntingtin spheroids and protofibrils as precursors in polyglutamine fibrilization. J Biol Chem 2002; 277: 41032–41037.
Ossato G, Digman MA, Aiken C, Lukacsovich T, Marsh JL, Gratton E . A two-step path to inclusion formation of huntingtin peptides revealed by number and brightness analysis. Biophys J 2010; 98: 3078–3085.
Rajan RS, Illing ME, Bence NF, Kopito RR . Specificity in intracellular protein aggregation and inclusion body formation. Proc Natl Acad Sci USA 2001; 98: 13060–13065.
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S . Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004; 431: 805–810.
Muchowski PJ . Protein misfolding, amyloid formation, and neurodegeneration: a critical role for molecular chaperones? Neuron 2002; 35: 9–12.
Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S et al. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet 2003; 12: 749–757.
Webb JL, Ravikumar B, Rubinsztein DC . Microtubule disruption inhibits autophagosome–lysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. Int J Biochem Cell Biol 2004; 36: 2541–2550.
Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J et al. Huntingtin localization in brains of normal and Huntington’s disease patients. Ann Neurol 1997; 42: 604–612.
Wang GH, Mitsui K, Kotliarova S, Yamashita A, Nagao Y, Tokuhiro S et al. Caspase activation during apoptotic cell death induced by expanded polyglutamine in N2a cells. NeuroReport 1999; 10: 2435–2438.
Pennuto M, Palazzolo I, Poletti A . Post-translational modifications of expanded polyglutamine proteins: impact on neurotoxicity. Hum Mol Genet 2009; 18: R40–47.
Gu X, Greiner ER, Mishra R, Kodali R, Osmand A, Finkbeiner S et al. Serines 13 and 16 are critical determinants of full-length human mutant huntingtin induced disease pathogenesis in HD mice. Neuron 2009; 64: 828–840.
Havel LS, Wang CE, Wade B, Huang B, Li S, Li XJ . Preferential accumulation of N-terminal mutant huntingtin in the nuclei of striatal neurons is regulated by phosphorylation. Hum Mol Genet 2011; 20: 1424–1437.
Harjes P, Wanker EE . The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci 2003; 28: 425–433.
Davranche A, Aviolat H, Zeder-Lutz G, Busso D, Altschuh D, Trottier Y et al. Huntingtin affinity for partners is not changed by polyglutamine length: aggregation itself triggers aberrant interactions. Hum Mol Genet 2011; 20: 2795–2806.
Chen S, Berthelier V, Yang W, Wetzel R . Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol 2001; 311: 173–182.
Wang Y, Meriin AB, Costello CE, Sherman MY . Characterization of proteins associated with polyglutamine aggregates: a novel approach towards isolation of aggregates from protein conformation disorders. Prion 2007; 1: 128–135.
Kitamura A, Kubota H, Pack CG, Matsumoto G, Hirayama S, Takahashi Y et al. Cytosolic chaperonin prevents polyglutamine toxicity with altering the aggregation state. Nat Cell Biol 2006; 8: 1163–1170.
Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, Morimoto RI . Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J 2004; 23: 4307–4318.
Schaffar G, Breuer P, Boteva R, Behrends C, Tzvetkov N, Strippel N et al. Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol Cell 2004; 15: 95–105.
Kim S, Nollen EA, Kitagawa K, Bindokas VP, Morimoto RI . Polyglutamine protein aggregates are dynamic. Nat Cell Biol 2002; 4: 826–831.
Mezey E, Dehejia A, Harta G, Papp MI, Polymeropoulos MH, Brownstein MJ . Alpha synuclein in neurodegenerative disorders: murderer or accomplice? Nat Med 1998; 4: 755–757.
Ueda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M et al. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci USA 1993; 90: 11282–11286.
Yoshimoto M, Iwai A, Kang D, Otero DA, Xia Y, Saitoh T . NACP the precursor protein of the non-amyloid beta/A4 protein (A beta) component of Alzheimer disease amyloid, binds A beta and stimulates A beta aggregation. Proc Natl Acad Sci USA 1995; 92: 9141–9145.
Charles V, Mezey E, Reddy PH, Dehejia A, Young TA, Polymeropoulos MH et al. Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models. Neurosci Lett 2000; 289: 29–32.
Corrochano S, Renna M, Carter S, Chrobot N, Kent R, Stewart M et al. Alpha-Synuclein levels modulate Huntington’s disease in mice. Hum Mol Genet 2012; 21: 485–494.
Tomas-Zapico C, Diez-Zaera M, Ferrer I, Gomez-Ramos P, Moran MA, Miras-Portugal MT et al. Alpha-Synuclein accumulates in huntingtin inclusions but forms independent filaments and its deficiency attenuates early phenotype in a mouse model of Huntington’s disease. Hum Mol Genet 2012; 21: 495–510.
Furlong RA, Narain Y, Rankin J, Wyttenbach A, Rubinsztein DC . Alpha-synuclein overexpression promotes aggregation of mutant huntingtin. Biochem J 2000; 346 (Part 3): 577–581.
Herrera F, Outeiro TF . Alpha-Synuclein modifies huntingtin aggregation in living cells. FEBS Lett 2012; 586: 7–12.
Caparros-Lefebvre D, Kerdraon O, Devos D, Dhaenens CM, Blum D, Maurage CA et al. Association of corticobasal degeneration and Huntington’s disease: can Tau aggregates protect Huntingtin toxicity? Mov Disord 2009; 24: 1089–1090.
Schwab C, Yu S, McGeer EG, McGeer PL . Optineurin in Huntington’s disease intranuclear inclusions. Neurosci Lett 2012; 506: 149–154.
Hortobagyi T, Troakes C, Nishimura AL, Vance C, van Swieten JC, Seelaar H et al. Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathol 2011; 121: 519–527.
Gong B, Lim MC, Wanderer J, Wyttenbach A, Morton AJ . Time-lapse analysis of aggregate formation in an inducible PC12 cell model of Huntington’s disease reveals time-dependent aggregate formation that transiently delays cell death. Brain Res Bull 2008; 75: 146–157.
Henshall TL, Tucker B, Lumsden AL, Nornes S, Lardelli MT, Richards RI . Selective neuronal requirement for huntingtin in the developing zebrafish. Hum Mol Genet 2009; 18: 4830–4842.
Soulet D, Cicchetti F . The role of immunity in Huntington’s disease. Mol Psychiatry 2011; 16: 889–902.
Turner C, Cooper JM, Schapira AH . Clinical correlates of mitochondrial function in Huntington’s disease muscle. Mov Disord 2007; 22: 1715–1721.
Chaturvedi RK, Adhihetty P, Shukla S, Hennessy T, Calingasan N, Yang L et al. Impaired PGC-1alpha function in muscle in Huntington’s disease. Hum Mol Genet 2009; 18: 3048–3065.
Orth M, Cooper JM, Bates GP, Schapira AH . Inclusion formation in Huntington’s disease R6/2 mouse muscle cultures. J Neurochem 2003; 87: 1–6.
Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, Zeisler J et al. Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell 1995; 81: 811–823.
Mazzola JL, Sirover MA . Reduction of glyceraldehyde-3-phosphate dehydrogenase activity in Alzheimer’s disease and in Huntington’s disease fibroblasts. J Neurochem 2001; 76: 442–449.
Squitieri F, Falleni A, Cannella M, Orobello S, Fulceri F, Lenzi P et al. Abnormal morphology of peripheral cell tissues from patients with Huntington disease. J Neural Transm 2010; 117: 77–83.
Manley K, Pugh J, Messer A . Instability of the CAG repeat in immortalized fibroblast cell cultures from Huntington’s disease transgenic mice. Brain Res 1999; 835: 74–79.
Maywood ES, Fraenkel E, McAllister CJ, Wood N, Reddy AB, Hastings MH et al. Disruption of peripheral circadian timekeeping in a mouse model of Huntington’s disease and its restoration by temporally scheduled feeding. J Neurosci 2010; 30: 10199–10204.
Chiang MC, Chern Y, Juo CG . The dysfunction of hepatic transcriptional factors in mice with Huntington’s disease. Biochim Biophys Acta 2011; 1812: 1111–1120.
Smith R, Bacos K, Fedele V, Soulet D, Walz HA, Obermuller S et al. Mutant huntingtin interacts with {beta}-tubulin and disrupts vesicular transport and insulin secretion. Hum Mol Genet 2009; 18: 3942–3954.
Cape A, Chen X, Wang CE, O’Neill A, Lin YF, He J et al. Loss of huntingtin-associated protein 1 impairs insulin secretion from pancreatic beta-cells. Cell Mol Life Sci 2012; 69: 1305–1317.
Metzler M, Helgason CD, Dragatsis I, Zhang T, Gan L, Pineault N et al. Huntingtin is required for normal hematopoiesis. Hum Mol Genet 2000; 9: 387–394.
Squitieri F, Cannella M, Sgarbi G, Maglione V, Falleni A, Lenzi P et al. Severe ultrastructural mitochondrial changes in lymphoblasts homozygous for Huntington disease mutation. Mech Ageing Dev 2006; 127: 217–220.
Maglione V, Cannella M, Gradini R, Cislaghi G, Squitieri F . Huntingtin fragmentation and increased caspase 3, 8 and 9 activities in lymphoblasts with heterozygous and homozygous Huntington’s disease mutation. Mech Ageing Dev 2006; 127: 213–216.
Squitieri F, Maglione V, Orobello S, Fornai F . Genotype-, aging-dependent abnormal caspase activity in Huntington disease blood cells. J Neural Transm 2011; 118: 1599–1607.
Blum D, Hourez R, Galas MC, Popoli P, Schiffmann SN . Adenosine receptors and Huntington’s disease: implications for pathogenesis and therapeutics. Lancet Neurol 2003; 2: 366–374.
Varani K, Abbracchio MP, Cannella M, Cislaghi G, Giallonardo P, Mariotti C et al. Aberrant A2A receptor function in peripheral blood cells in Huntington’s disease. FASEB J 2003; 17: 2148–2150.
Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ . Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol 2005; 171: 1001–1012.
Bradford J, Shin JY, Roberts M, Wang CE, Li XJ, Li S . Expression of mutant huntingtin in mouse brain astrocytes causes age-dependent neurological symptoms. Proc Natl Acad Sci USA 2009; 106: 22480–22485.
Bradford J, Shin JY, Roberts M, Wang CE, Sheng G, Li S et al. Mutant huntingtin in glial cells exacerbates neurological symptoms of Huntington disease mice. J Biol Chem 2010; 285: 10653–10661.
Chou SY, Weng JY, Lai HL, Liao F, Sun SH, Tu PH et al. Expanded-polyglutamine huntingtin protein suppresses the secretion and production of a chemokine (CCL5/RANTES) by astrocytes. J Neurosci 2008; 28: 3277–3290.
Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum Mol Genet 2010; 19: 3053–3067.
Chen LL, Wu JC, Wang LH, Wang J, Qin ZH, Difiglia M et al. Rapamycin prevents the mutant huntingtin-suppressed GLT-1 expression in cultured astrocytes. Acta Pharmacol Sin 2012; 33: 385–392.
Wang L, Lin F, Wang J, Wu J, Han R, Zhu L et al. Expression of mutant N-terminal huntingtin fragment (htt552-100Q) in astrocytes suppresses the secretion of BDNF. Brain Res 2012; 1449: 69–82.
Anderson AN, Pavese N, Edison P, Tai YF, Hammers A, Gerhard A et al. A systematic comparison of kinetic modelling methods generating parametric maps for [(11)C]-(R)-PK11195. Neuroimage 2007; 36: 28–37.
Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain 2007; 130 (Part 7): 1759–1766.
Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ et al. Imaging microglial activation in Huntington’s disease. Brain Res Bull 2007; 72: 148–151.
Silvestroni A, Faull RL, Strand AD, Moller T . Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. NeuroReport 2009; 20: 1098–1103.
Bjorkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med 2008; 205: 1869–1877.
Giorgini F, Moller T, Kwan W, Zwilling D, Wacker JL, Hong S et al. Histone deacetylase inhibition modulates kynurenine pathway activation in yeast, microglia, and mice expressing a mutant huntingtin fragment. J Biol Chem 2008; 283: 7390–7400.
Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 1997; 90: 537–548.
Kraft AD, Kaltenbach LS, Lo DC, Harry GJ . Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiol Aging 2012; 33: 621 e617–633.
Ma L, Morton AJ, Nicholson LF . Microglia density decreases with age in a mouse model of Huntington’s disease. Glia 2003; 43: 274–280.
Simmons DA, Casale M, Alcon B, Pham N, Narayan N, Lynch G . Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington’s disease. Glia 2007; 55: 1074–1084.
Tydlacka S, Wang CE, Wang X, Li S, Li XJ . Differential activities of the ubiquitin–proteasome system in neurons versus glia may account for the preferential accumulation of misfolded proteins in neurons. J Neurosci 2008; 28: 13285–13295.
Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A et al. Caspase signalling controls microglia activation and neurotoxicity. Nature 2011; 472: 319–324.
Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G et al. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell 2006; 125: 1179–1191.
Valenza M, Cattaneo E . Cholesterol dysfunction in neurodegenerative diseases: is Huntington’s disease in the list? Prog Neurobiol 2006; 80: 165–176.
Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D . Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006; 127: 59–69.
Xiang Z, Valenza M, Cui L, Leoni V, Jeong HK, Brilli E et al. Peroxisome-proliferator-activated receptor gamma coactivator 1 alpha contributes to dysmyelination in experimental models of Huntington’s disease. J Neurosci 2011; 31: 9544–9553.
Browne SE, Beal MF . Oxidative damage in Huntington’s disease pathogenesis. Antioxid Redox Signal 2006; 8: 2061–2073.
St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jager S et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006; 127: 397–408.
Camnasio S, Carri AD, Lombardo A, Grad I, Mariotti C, Castucci A et al. The first reported generation of several induced pluripotent stem cell lines from homozygous and heterozygous Huntington’s disease patients demonstrates mutation related enhanced lysosomal activity. Neurobiol Dis 2012; 46: 41–51.
Castiglioni V, Onorati M, Rochon C, Cattaneo E . Induced pluripotent stem cell lines from Huntington’s disease mice undergo neuronal differentiation while showing alterations in the lysosomal pathway. Neurobiol Dis 2012; 46: 30–40.
Ritch JJ, Valencia A, Alexander J, Sapp E, Gatune L, Sangrey GR et al. Multiple phenotypes in Huntington disease mouse neural stem cells. Mol Cell Neurosci 2012; 50: 70–81.
Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A et al. Disease-specific induced pluripotent stem cells. Cell 2008; 134: 877–886.
Ho LW, Brown R, Maxwell M, Wyttenbach A, Rubinsztein DC . Wild type Huntingtin reduces the cellular toxicity of mutant Huntingtin in mammalian cell models of Huntington’s disease. J Med Genet 2001; 38: 450–452.
Song C, Perides G, Liu YF . Expression of full-length polyglutamine-expanded Huntingtin disrupts growth factor receptor signaling in rat pheochromocytoma (PC12) cells. J Biol Chem 2002; 277: 6703–6707.
Colby DW, Cassady JP, Lin GC, Ingram VM, Wittrup KD . Stochastic kinetics of intracellular huntingtin aggregate formation. Nat Chem Biol 2006; 2: 319–323.
Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O . Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet 2008; 17: 345–356.
Ye C, Zhang Y, Wang W, Wang J, Li H . Inhibition of neurite outgrowth and promotion of cell death by cytoplasmic soluble mutant huntingtin stably transfected in mouse neuroblastoma cells. Neurosci Lett 2008; 442: 63–68.
Olshina MA, Angley LM, Ramdzan YM, Tang J, Bailey MF, Hill AF et al. Tracking mutant huntingtin aggregation kinetics in cells reveals three major populations that include an invariant oligomer pool. J Biol Chem 2010; 285: 21807–21816.
Wang HQ, Xu YX, Zhao XY, Zhao H, Yan J, Sun XB et al. Overexpression of F(0)F(1)-ATP synthase alpha suppresses mutant huntingtin aggregation and toxicity in vitro. Biochem Biophys Res Commun 2009; 390: 1294–1298.
Dong G, Ferguson JM, Duling AJ, Nicholas RG, Zhang D, Rezvani K et al. Modeling pathogenesis of Huntington's disease with inducible neuroprogenitor cells. Cell Mol Neurobiol 2011; 31: 737–747.
Miller J, Arrasate M, Shaby BA, Mitra S, Masliah E, Finkbeiner S . Quantitative relationships between huntingtin levels, polyglutamine length, inclusion body formation, and neuronal death provide novel insight into huntington's disease molecular pathogenesis. J Neurosci 2010; 30: 10541–10550.
Lunkes A, Mandel JL . A cellular model that recapitulates major pathogenic steps of Huntington’s disease. Hum Mol Genet 1998; 7: 1355–1361.
Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J et al. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet 2002; 11: 1953–1965.
Igarashi S, Morita H, Bennett KM, Tanaka Y, Engelender S, Peters MF et al. Inducible PC12 cell model of Huntington’s disease shows toxicity and decreased histone acetylation. Neuroreport 2003; 14: 565–568.
Aiken CT, Tobin AJ, Schweitzer ES . A cell-based screen for drugs to treat Huntington's disease. Neurobiol Dis 2004; 16: 546–555.
Weiss A, Roscic A, Paganetti P . Inducible mutant huntingtin expression in HN10 cells reproduces Huntington’s disease-like neuronal dysfunction. Mol Neurodegener 2009; 4: 11.
Podolsky S, Leopold NA . Abnormal glucose tolerance and arginine tolerance tests in Huntington’s disease. Gerontology 1977; 23: 55–63.
Moffitt H, McPhail GD, Woodman B, Hobbs C, Bates GP . Formation of polyglutamine inclusions in a wide range of non-CNS tissues in the HdhQ150 knock-in mouse model of Huntington's disease. PLoS One 2009; 4: e8025.
Garewal HS, Leibovitz A, Sampliner RE, Ramsey L, Hendrix MJ, Sloan D . Tissue culture of epithelial cells from esophageal specialized columnar epithelium (Barrett’s esophagus). Dig Dis Sci 1992; 37: 532–536.
Ribchester RR, Thomson D, Wood NI, Hinks T, Gillingwater TH, Wishart TM et al. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington's disease mutation. Eur J Neurosci 2004; 20: 3092–3114.
Ciammola A, Sassone J, Alberti L, Meola G, Mancinelli E, Russo MA et al. Increased apoptosis, Huntingtin inclusions and altered differentiation in muscle cell cultures from Huntington’s disease subjects. Cell Death Differ 2006; 13: 2068–2078.
Boesgaard TW, Nielsen TT, Josefsen K, Hansen T, Jorgensen T, Pedersen O et al. Huntington’s disease does not appear to increase the risk of diabetes mellitus. J Neuroendocrinol 2009; 21: 770–776.
Van Raamsdonk JM, Murphy Z, Selva DM, Hamidizadeh R, Pearson J, Petersen A et al. Testicular degeneration in Huntington disease. Neurobiol Dis 2007a; 26: 512–520.
Van Raamsdonk JM, Metzler M, Slow E, Pearson J, Schwab C, Carroll J et al. Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol Dis 2007b; 26: 189–200.
Almeida S, Sarmento-Ribeiro AB, Januario C, Rego AC, Oliveira CR . Evidence of apoptosis and mitochondrial abnormalities in peripheral blood cells of Huntington’s disease patients. Biochem Biophys Res Commun 2008; 374: 599–603.
Pattison JS, Sanbe A, Maloyan A, Osinska H, Klevitsky R, Robbins J . Cardiomyocyte expression of a polyglutamine preamyloid oligomer causes heart failure. Circulation 2008; 117: 2743–2751.
Sathasivam K, Hobbs C, Turmaine M, Mangiarini L, Mahal A, Bertaux F et al. Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet 1999; 8: 813–822.
Mihm MJ, Amann DM, Schanbacher BL, Altschuld RA, Bauer JA, Hoyt KR . Cardiac dysfunction in the R6/2 mouse model of Huntington’s disease. Neurobiol Dis 2007; 25: 297–308.
Yoshida Y, Yamanaka S . Recent stem cell advances: induced pluripotent stem cells for disease modeling and stem cell-based regeneration. Circulation 2010; 122: 80–87.
Sreejit P, Kumar S, Verma RS . An improved protocol for primary culture of cardiomyocyte from neonatal mice. In Vitro Cell Dev Biol Anim 2008; 44: 45–50.
Acknowledgements
The authors would like to acknowledge the support of the Huntington Society Canada and the International Organization of Glutaric Acidemia (IOGA) in part to Francesca Cicchetti. Giulia Cisbani was supported by PhD recruitment scholarship from Université Laval. The authors also wish to thank Mr. Gilles Chabot for artwork and Richard Poulin for editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by D Bano
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Cisbani, G., Cicchetti, F. An in vitro perspective on the molecular mechanisms underlying mutant huntingtin protein toxicity. Cell Death Dis 3, e382 (2012). https://doi.org/10.1038/cddis.2012.121
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cddis.2012.121
Keywords
This article is cited by
-
Spatiotemporal analysis of soluble aggregates and autophagy markers in the R6/2 mouse model
Scientific Reports (2021)
-
Modulation of in vitro Brain Endothelium by Mechanical Trauma: Structural and Functional Restoration by Poloxamer 188
Scientific Reports (2020)
-
Discovery of a potent small molecule inhibiting Huntington’s disease (HD) pathogenesis via targeting CAG repeats RNA and Poly Q protein
Scientific Reports (2019)
-
Mutant Huntingtin Causes a Selective Decrease in the Expression of Synaptic Vesicle Protein 2C
Neuroscience Bulletin (2018)
-
The dynamics of early-state transcriptional changes and aggregate formation in a Huntington’s disease cell model
BMC Genomics (2017)
