
Abstract
Huntington disease (HD) is a hereditary neurodegenerative disorder caused by mutant huntingtin (mHTT). Phosphorylation at serine-421 (pS421) of mHTT has been shown to be neuroprotective in cellular and rodent models. However, the genetic context of these models differs from that of HD patients. Here we employed human pluripotent stem cells (hiPSCs), which express endogenous full-length mHTT. Using genome editing, we generated isogenic hiPSC lines in which the S421 site in mHTT has been mutated into a phospho-mimetic aspartic acid (S421D) or phospho-resistant alanine (S421A). We observed that S421D, rather than S421A, confers neuroprotection in hiPSC-derived neural cells. Although we observed no effect of S421D on mHTT clearance or axonal transport, two aspects previously reported to be impacted by phosphorylation of mHTT at S421, our analysis revealed modulation of several aspects of mitochondrial form and function. These include mitochondrial surface area, volume, and counts, as well as improved mitochondrial membrane potential and oxidative phosphorylation. Our study validates the protective role of pS421 on mHTT and highlights a facet of the relationship between mHTT and mitochondrial changes in the context of human physiology with potential relevance to the pathogenesis of HD.
Similar content being viewed by others


Introduction
Huntington disease (HD), a devastating hereditary neurodegenerative disorder, is caused by an expansion of a CAG trinucleotide repeat that encodes a polyglutamine (polyQ) tract in the huntingtin (HTT) gene. HTT is a ubiquitously expressed 350 kDa protein that is found in different subcellular compartments, including the nucleus, mitochondria, the Golgi complex, microtubules, and vesicular structures in neurites and synapses. Proposed to act as a scaffold1, HTT interacts with a large number of proteins. Posttranslational modifications (PTMs) of HTT are thought to be key determinants of HTT’s conformation as well as its ability to associate with its interacting partners. These PTMs have been shown to include phosphorylation, acetylation, SUMOlyation, proteolytic cleavage, palmitoylation, and ubiquitination2,3. The toxicity of the expanded polyQ tract is postulated to result, at least in part, from disrupted interactions with molecular partners as a consequence of altered PTMs of HTT.
Converging lines of evidence support a role for phospho-S421 (pS421) levels in modulating mutant HTT (mHTT) toxicity. The pro-survival signaling protein Akt is one of the kinases shown to phosphorylate HTT on S4211,4. pS421-HTT levels show a regional distribution that is inversely correlated with neuronal atrophy in HD, with the highest levels in the cerebellum, less in the cortex, and least in the striatum5. pS421-HTT levels decline steadily over time in the striatum of YAC128 HD mice6. A number of cellular defects have been linked to the reduced levels of pS421-HTT, including intracellular transport defects7, increased proteolysis and nuclear accumulation of HTT fragments8, and diminished neuronal survival6,9.
Although previous studies have explored the impact of PTMs on mHTT function and toxicity, the interpretation of the findings from these studies is confounded by caveats that include the following: (a) the use of truncated N-terminal fragments of mHTT that comprise only 67 to ~700 amino acids (aa) of the 3144 aa full-length protein; (b) the use of non-endogenous promoters typically resulting in expression levels that are several fold higher than the endogenous mHTT levels; and (c) the use of non-neuronal cell lines such as HEK293 or HeLa cells, or rodent-derived neuronal cell lines such as mouse Neuro2a cells10,11,12,13. Therefore, understanding the importance of PTMs in the context of human pathology remains a major gap in our knowledge of the significance of HTT PTMs specifically and the pathogenesis of HD in general.
Here we sought to examine the impact of pS421 of HTT on expanded polyQ-related phenotypes in human induced pluripotent stem cell (hiPSC)-derived neural cells. Using a TALEN (transcription activator-like effector nuclease)-based genomic editing approach, we mutated polyQ-expanded HTT at S421 to create a variant that is resistant to phosphorylation at S421 (a serine to alanine substitution, S421A) or one that mimics phosphorylation (a serine to aspartic acid substitution, S421D). The functional consequences of the phosphorylation-competent polyQ-expanded HTT (S421S) were compared to phosphorylation-resistant polyQ-expanded HTT (S421A) and pseudo-phosphorylated polyQ-expanded HTT (S421D) in hiPSC-derived neural cells. We observed that S421D, rather than S421A, confers neuroprotection against mHTT toxicity in hiPSC-derived neural cells. We further found that this neuroprotection was associated with modulation of several aspects of mitochondrial form and function including mitochondrial surface area, volume, and counts, as well as improved mitochondrial membrane potential and oxidative phosphorylation. Our study highlights a facet of the relationship between mHTT and mitochondrial changes in the context of human physiology with potential relevance to the pathogenesis of HD. Furthermore, the approach we describe here can be applied widely to investigate the effect of other PTMs on neurodegenerative disease phenotypes in a genetically faithful and physiologically relevant human context.
Materials and methods
Cloning of constructs
S421-T1 and S421-T2 TALENs were cloned into pCDNA4 vector by a single step of the 4 × 4 Golden Gate assembly protocol using a premade tetramer TALE repeat library by the Biological Resource Centre, A*STAR, Singapore. Subsequently, S421-T1 and S421-T2 TALENs were cloned into pTAL-GFP (Addgene, #43858) and pTAL-RFP (Addgene, #43859) vector using BamHI and Xbal restriction enzymes, respectively. Single-stranded oligodeoxynucleotides (ssODNs) donors for S421 site targeting were synthesized by Sigma-Aldrich. S421-T1 and S421-T2 TALEN-binding sequence and ssODN sequence are listed in Supplementary Table S1.
Cell culture
HEK293 cells for TALENs activity test were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). K562 cells for ssODNs donor test were cultured in Iscove’s modified Dulbecco’s medium supplemented with 10% FBS. CAG180 hiPSCs (ND36999) and CAG33 hiPSCs (ND36997) were obtained from the NINDS hiPSC Repository at Coriell Institute. HD-C hiPSCs are isogenic lines established by correction of mutant CAG180 repeats with 18 CAG repeats as described previously14. All hiPSCs were cultured on Matrigel (BD Biosciences)-coated plates with mTesR-1 medium (Stemcell Technologies, #05850).
Differentiation of hiPSCs
NPC differentiation
hiPSCs were induced into neural progenitor cells (NPCs) according to a previously published protocol15. Briefly, hiPSCs at ~20% confluence were treated with N2B27 media (DMEM-F12/Neural Basal medium 1 : 1 with 1% N2, 2% B27, 1% pen/strep/glutamine, 10 ng/mL hLIF, and 5 μg/mL bovine serum albumin) containing 3 μM CHIR99021 (Tocris), 2 μM SB431542 (Sigma), and 0.1 μM compound E (Millipore) for the first 7 days. The culture was then split 1 : 3 for the next six passages using Accutase without compound E on Matrigel-coated plates. Cells on passage 3 and passage 4 were used for experiments.
Forebrain neuronal differentiation
hiPSCs were differentiated into forebrain neurons using our previously published protocol14,16. Briefly, NPCs were induced in N2B27 medium supplemented with certain small molecules and growth factors (GFs) for 15 days. For final neuronal differentiation, the cells were cultured in N2B27 medium supplemented with brain-derived neurotrophic factor (BDNF) (20 ng/ml), glial cell line-derived neurotrophic factor (GDNF) (20 ng/ml), dbcAMP (0.5 mM), and Ascorbic Acid (0.2 mM). Half the medium was replaced with fresh medium every 3–4 days for the terminally differentiated neurons.
Cortical neuron differentiation
Differentiation of hiPSCs into cortical neurons was performed as previously described17. The cortical neurons on Day 53 were dissociated and re-plated on glass-bottom dishes for mitochondrial axonal transport analysis.
YOYO-1 cell death assay
NPCs were dissociated into single cells using Accutase and seeded onto Matrigel-coated 96-well plate and cultured overnight. Next day, cells were first treated with 200 µM tert-butyl hydroperoxide (TBHP) for 2 h and switched to fresh NPC medium containing 100 nM YOYO-1 (Thermo Fisher Scientific, catalog number Y3601). Imaging was performed for 24 h using the Incucyte Zoom system (Essen Bioscience). YOYO-1 expression levels on 24 h were normalized to the 0 h expression levels for each well and the data from two individual clones per genotype were combined together for statistical analysis.
MitoTracker staining and flow analysis
NPCs were stained with 100 nM MitoTracker Green FM (Invitrogen, M7514) and 100 nM MitoTracker Red CMXRos (Invitrogen, M7512) for 15 min at 37 °C, according to the manufacturer’s instructions. The stained cells were run on CytoFLEX flow cytometer (Beckman Coulter) and the mean fluorescence intensity were used for analysis.
Mitochondrial respiration analysis
The NPCs were plated on the 96-well assay plate at a density of 150,000 cells/well 1 day before assay. To measure the mitochondrial respiration under oxidative stress conditions, cells were treated with 200 µM TBHP for 2 h and switched to DMEM/F12 medium for another 2 h before assay. The assay was carried out using a Seahorse XF96 Extracellular Flux Analyzer followed the instructions of Seahorse XF Cell Mito Stress Test Kit (Seahorse Bioscience). The readings were normalized to total protein amount measured by Bradford assay and data were analyzed using the Seahorse Wave software.
RNA-Seq analysis
RNA was extracted from cells using a RNeasy plus mini kit (Qiagen), according to the manufacturer’s instructions. Subsequent library preparation and paired-end 100 bp sequencing and 30 M reads/per sample using Illumina HiSeq4000-PE150 were performed by Novogene (Hong Kong). Gene set enrichment analysis (GSEA) was used to test whether the gene expression signatures of pS421-mHTT were enriched for Kyoto Encyclopedia of Genes and Genomes, Reactome, and Gene Ontology Biological Process pathways18,19,20. The RNA sequencing (RNA-Seq) data were deposited into Sequence Read Archive (SRA) with the Bioproject ID of PRJNA474894. See Supplementary Information for the details of the protocol.
Measurement of mitochondrial morphology
Cells were fixed and immunostained with anti-TOM20 (1 : 500; Proteintech, 11802–1-AP) primary antibodies and Alexa Fluor 568-conjugated secondary antibodies to visualize the mitochondrial network. A DeltaVision OMX v4 Blaze microscope (GE Healthcare, Issaquah, WA, USA) was used for acquisition of widefield-deconvolved images of individual NPCs. Images were acquired at a z-spacing of 0.125 µm as previously described21. To measure mitochondrial morphological parameters, the deconvolved widefield images were imported into IMARIS (Bitplane) for three-dimensional (3D) reconstruction. Raw data points of each mitochondrion detected were exported for statistical analysis. See Supplementary Information for the details of the protocol.
The detailed methods for nucleofection of hiPSCs, screening targeted clones, allele-specific PCR, fragment sizing analysis, RNA isolation, cDNA synthesis and quantitative PCR, CellTiter-Glo® luminescent cell viability assay, GF withdrawal assay, measurement of mitochondrial morphology, immunoblotting, immunofluorescence staining, and RNA-Seq analysis are found in the Supplementary Materials and Methods.
Statistics
For experiments with hiPSC-derived neural cells, each biological replicate was derived from an independently differentiated clone and subsequent assessment. All statistical analyses were performed in GraphPad Prism 7 software. One-way analysis of variance was used to test for differences across different groups, followed by a Fisher’s least significant difference post hoc multiple-comparison test to test for specific differences between groups. Otherwise, for comparisons of the mean between two groups, Student’s t-test was applied. All the data were shown as mean ± SEM and p-values were considered as follows: ns, no significance; *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
Generation of isogenic S421A/D-mHTT hiPSCs
A published CAG180 HD hiPSC line (Coriell, ND36999), demonstrating certain distinct HD phenotypes14,22, was chosen as the parental cell line to generate isogenic S421A and S421D HD hiPSC lines. We designed a pair of TALENs, which could bind to the DNA sequences around the S421 site (Fig. 1a; S421-T1/2) with very few off-target sites predicted by TAL Effector Nucleotide Targeter 2.0 (Table S2). S421-T1 and S421-T2 TALENs demonstrated efficient cleavage activity assessed by surveyor assay in HEK293 cells (Fig. 1b). To mutate the serine at S421 site into phospho-mimetic aspartic acid (S421D) or phospho-resistant alanine (S421A), 90 bp ssODNs were used as donors. The ssODNs contained two nucleotides “GC/GA” mutations for converting “S” into “A” or “D” at the S421 site, respectively, and one additional synonymous “C” nucleotide mutation to introduce a BglI restriction enzyme site to facilitate targeted clone screening (Fig. 1c). The homology-directed repair by ssODNs and screening strategy were validated in human K562 cells (Fig. 1d). As illustrated, the PCR products after BglI digestion were predicted to result in one band for non-targeted clones, two lower bands for targeted homozygous clones, and three bands for heterozygous or mixed clones (Fig. 1e). Using this strategy, we electroporated green fluorescent protein (GFP)-tagged S421-T1, red fluorescent protein (RFP)-tagged S421-T2 TALENs, and ssODNs into CAG180 hiPSCs. The GFP(+)/RFP(+) hiPSCs were sorted into 96-well plates for clone expansion and PCR screening (Fig. 1f). The targeting efficiency was around 3% in CAG180 hiPSCs by PCR screening (Fig. 1g). To determine the targeted allele, we employed allele-specific PCR using a known single-nucleotide polymorphism upstream of S421 site on the mutant allele in CAG180 line (Fig. 1h). Moreover, the clones that had undergone the whole targeting process without modification on S421 sites were used as control clones (referred to as S421S) and clones only targeted on the mutant allele were named as S421A or S421D. All candidate subclones of S421S, S421A, and S421D iPSCs for further validation were heterozygous and at least two subclones per genotype groups were used for subsequent experiments.

a TALEN-binding sequences flanking the S421 codon in HTT exon 9. b TALEN cleavage activity as assessed by Surveyor assay. c ssODN donor DNA with BglI restriction enzyme site for screening. d Verification of targeting and screening strategy in K562 cells. e Expected restriction fragment pattern with BglI screening. f HTT Exon 9 PCR followed by BglI screening identifies a number of targeted CAG180 HD hiPSC clones (black arrows); example shown is for S421(A) targeting; red arrows represent mixed clones. g Summary of targeting efficiency. h Phasing of targeted allele by allele-specific PCR followed by Sanger sequencing.
We then examined the pluripotency of the targeted clones by OCT4 staining and mRNA expression of pluripotent marker OCT4 and LIN28. All lines had similar expression levels of OCT4 and LIN28 compared with the parental CAG180 HD hiPSC line (Supplementary Fig. S1a, b). Karyotyping and G-banding analysis demonstrated that all clones maintained a normal 46, XY karyotype (Supplementary Fig. S1c) and they demonstrated similar CAG expansion size and pattern via CAG fragment sizing analysis (Supplementary Fig. S1d).
S421 phosphorylation status influences susceptibility to cell death
To validate the neuroprotective role of S421D in iPSC-derived neural cells, we first examined the susceptibility of NPCs derived from S421S, S421A, and S421D HD hiPSCs to oxidative stress. NPCs were treated with 200 µM TBHP for 2 h, after which cells were cultured for another 24 h before being assayed. YOYO-1, a green fluorescence dye that stains the nuclear DNA of cells that have lost plasma membrane integrity23, was used to quantify cell death. S421D NPCs showed less cell death after TBHP treatment compared to S421S and S421A NPCs (Fig. 2a). Consistently, cell viability of S421D NPCs after TBHP treatment as measured by CellTiter-Glo assay was improved as compared to that of the S421S and S421A lines (Fig. 2b).

a TBHP-induced cell death in neural progenitor cells (NPCs) measured using the YOYO-1 cell death assay. n = 3 independent biological replicates (BRs). b Cell viability of neural progenitor cells following TBHP treatment determined using the ATP assay. n = 3 BRs. a, b n.s., no significance and **p < 0.01 was determined by one-way ANOVA analysis followed by a Fisher’s LSD post hoc multiple-comparison test. c Schematic of neuronal death assay after growth factor withdrawal. d TUNEL immunofluorescence images showing cell death in S421S, S421A, and S421D neurons following growth factor withdrawal. e Quantification of cell death as measured by TUNEL staining in S421S, S421A, and S421D neurons following growth factor withdrawal. n = 6 for 2 clones per genotype with 3 BRs. Two subclones were used per genotype group. Values shown as mean ± SEM and n.s., no significance, *p < 0.05 and ***p < 0.001 for shown comparisons, and $p < 0.05 and $$p < 0.01 relative to S421S-Control was determined by unpaired t-test.
We next sought to determine whether the S421 phosphorylation status would influence the susceptibility of post-mitotic HD neurons to GF withdrawal, as we previously observed that HD neurons were more susceptible to cell death after GF withdrawal compared with corrected isogenic control neurons14. Post-mitotic neurons differentiated from hiPSCs on Day 40 were used for the GF withdrawal assay (Fig. 2c and Supplementary Fig. S2). Under normal culture conditions with GF, the S421A showed an increased cell death, whereas S421D neurons had less cell death compared with S421S neurons (Fig. 2d, e). Moreover, GF withdrawal resulted in increased cell death of S421S and S421A neurons, in contrast to S421D neurons, which did not show any increase in cell death following GF withdrawal (Fig. 2d, e).
In summary, we observed that S421D, the pseudo-phosphorylated status, rather than phospho-resistant S421A, protected HD hiPSC-derived NPCs and neurons against cell death caused by oxidative stress and GF withdrawal, respectively.
S421 phosphorylation status does not affect proteasome-dependent clearance of mHTT, axonal transport, or neural rosette formation
A recent study suggested that phosphorylation at S421 may affect proteasome-dependent turnover of mHTT24. We examined HTT levels and found no significant differences in the expression levels of total HTT and mHTT protein across S421S, S421A, and S421D hiPSC lines (Fig. 3a, b), as well as NPCs (Fig. 3c–e) under normal culture conditions. After 24 h treatment with the proteasomal inhibitor Lactacystin (1 μM) or the lysosomal inhibitors ammonium chloride (NH4Cl, 20 nM) and Leupeptin (100 μM), we only observed increased mHTT in S421S NPCs treated with lysosomal inhibitors compared with the control group, but no changes in total HTT or wild-type (WT) HTT levels (Fig. 3c, f). This suggests the lysosomal pathway is an important pathway for mHTT degradation, and that S421A and S421D might increase lysosomal-dependent turnover of mHTT in our iPSC model. Furthermore, proteasomal and lysosomal inhibitors had no effect on total, WT, or mHTT levels in the S421A or S421D HD lines (Fig. 3c–e).

a, b Expression levels of HTT in hiPSCs by immunoblotting (a) and quantification of total HTT, WT HTT, and mutant HTT (b). HD-C is a corrected isogenic line for parental CAG180 HD iPSC line. c–f HTT clearance assay in NPCs derived from isogenic S421S, S421A, and S421D HD hiPSCs. C, Control; Lac, 1 μM Lactacystin; N + L, 200 nM NH4Cl and 10 μM Leupeptin. Representative immunoblotting image (c) and quantification of total HTT (d), WT HTT (e), and mutant HTT (f). Values shown as mean ± SEM and *p < 0.05 for shown comparisons was determined by one-way ANOVA followed by a Fisher’s LSD post hoc multiple-comparison test. n = 6 for 2 clones per group from 3 BRs. g Schematic of mitochondrial transport analysis. h Total vesicle moving distance in 5 min, n = 76 for S421S, 70 for S421A, and 77 for S421D. i Stationary events per vesicle in 5 min, n = 76 for S421S, 70 for S421A, and 77 for S421D. j Anterograde velocity, n = 72 for S421S, 70 for S421A, and 74 for S421D. Retrograde velocity, n = 76 for S421S, 70 for S421A, and 77 for S421D. Values were from three BRs and shown as mean ± SEM, and n.s., no significance.
Several lines of evidence demonstrated that mHTT affects mitochondrial axonal transport in HD25,26,27. Here we tested whether mitochondrial trafficking was influenced by phosphorylation status at S421 using cortical neurons differentiated from S421S, S421A, and S421D HD hiPSCs (Fig. 3g). However, we did not find significant differences among S421S, S421A, and S421D neurons in the total axonal distance traveled in a 5 min interval (Fig. 3h), stationary events (Fig. 3i), and anterograde and retrograde velocity (Fig. 3j) of mitochondria.
Finally, we examined whether S421 phosphorylation status affects neural rosette formation. We have previously shown that the ability of hiPSCs to form neural rosettes is impaired by mHTT14. We performed the neural rosette formation assay on the S421S, S421A, and S421D hiPSC lines. We found that neither S421A nor S421D had an apparent effect on the deficits in neural rosette formation observed in HD hiPSCs (Supplementary Fig. S3).
Analysis of differential gene expression in S421S and S421A/D NPCs
To gain insight into the cellular processes that might underlie the neuroprotection conferred by S421D relative to S421S and S421A, we performed RNA-Seq analysis on S421S, S421A, and S421D NPCs. For each genotype, two replicates from two individual clones were used for analysis of differentially expressed genes (DEGs). We identified a number of transcriptional gene expression changes associated with the S421 phosphorylation status (Fig. 4a). There were 1493 downregulated DEGs and 1129 upregulated genes in S421A vs. S421S NPCs, and 1465 downregulated genes and 1250 upregulated genes in S421D vs. S421S NPCs (Fig. 4b). GSEA results show that DEG-related mitochondrial translation, respiratory electron transport, and oxidative phosphorylation pathways were significantly enriched in both S421A and S421D gene expression signatures (Fig. 4c). The S421D gene expression signature was also significantly enriched for Huntington’s disease, response to oxygen levels, axon guidance, and regulation of cell activation/growth-related Gene Ontology (GO) terms (Fig. 4c). A clustered heatmap of the top 15 downregulated DEGs and top 15 upregulated DEGs is shown in Fig. 4d.

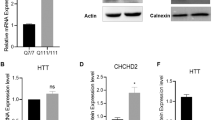
a Volcano plot of S421A vs. S421S and S421D vs. S421S (−log10(p-value) vs. log2FC), highlighting DEGs with nominal p < 0.05. b Number of up and down DEGs of S421A vs. S421S, and S421D vs. S421S with nominal p < 0.05. c GSEA. d Gene Venn analysis of specific DEGs of S421D vs. S421S with adjusted p < 0.05. d Heatmap of expression level for the top 30 genes only differentially expressed in S421D vs. S421S not in S421A vs. S421S. Heatmap represent four samples (two biological replicates from two independent clones) per genotype. e Expression levels of CHCHD2 in NPCs measured by qRT-PCR; Values shown as mean ± SEM; n = 3 BRs. f Expression levels of CHCHD2 in hiPSCs and NPCs measured by immunoblotting and relative quantification. g Values shown as mean ± SEM; n = 4 BRs for iPSCs and n = 3 BRs for NPCs (two subclones were used per genotype).
CHCHD2 is one of the top DEGs identified in our analysis of S421D vs. S421S NPCs and has been previously reported to be highly expressed in HD cells and tissues14,28,29. Therefore, we sought to further validate CHCHD2 expression in S421S and S421A/D hiPSCs and NPCs. Consistent with the RNA-Seq results, both mRNA levels of CHCHD2 (Fig. 4e) and CHCHD2 protein expression (Fig. 4f, g) were markedly higher in S421S and S421A cells compared with the isogenic control and S421D groups.
S421 phosphorylation status affects mitochondrial morphology in NPCs
Animal and cell models of HD have provided compelling evidence that mitochondrial morphology and function are affected in HD25,30. Given that our transcriptional analysis suggested that mitochondrial pathways are influenced by S421 phosphorylation status, we sought to examine whether mitochondria are affected in S421D and S421A compared with S421S neural cells. We first imaged NPCs stained with TOM20, a mitochondrial marker, using high-resolution microscopy and then applied 3D reconstruction using the IMARIS software to quantify aspects of mitochondrial morphology. In support of the transcriptional analysis, we found a number of measures of mitochondrial morphology to be altered in S421D cells compared with S421S and S421A. S421D cells presented less mitochondrial surface area and volume, increased surface area to volume ratio, and more mitochondrial counts compared with S421S and S421A cells (Fig. 5a–e). Second, using established mitochondrial assays with fluorescent dyes in live cells31,32, we also observed increased total mitochondrial content in S421D NPCs (as measured by Mitotracker Green FM) when compared with S421S and S421A groups (Fig. 5f). These findings suggest that S421 phosphorylation status of mHTT affects processes that influence mitochondrial morphology.

a Representative images of mitochondrial morphology in HD NPCs; Tom-20, orange; DNA, blue; scale bar = 5 μm. b–e Metrics of mitochondrial morphology in HD NPCs: surface area (b); volume (c); surface area to volume ratio (d); mitochondrial counts (e); n = 5599 mitochondria for S421S, 5463 mitochondria for S421A, 11,710 mitochondria for S421D from 2 different BRs (2 subclones were used per genotype). Values shown as mean ± SEM, n.s., no significance, and ***p < 0.001 was determined by Kruskal–Wallis one-way ANOVA with Dunn’s multiple comparisons test. f NPCs under basal culture conditions were stained with total mitochondrial marker Mitotracker Green FM; n = 6 for 2 clones per genotype with 3 BRs.
S421 phosphorylation status affects mitochondrial function in NPCs
To explore whether S421 phosphorylation status affects mitochondrial function, we first used Mitotracker Red CMXRos, a red fluorescent dye that stains mitochondria in live cells, to measure the amount of functional mitochondria. Although there was no difference between S421S, S421A, and S421D NPCs at baseline (Fig. 6a), the functional mitochondria content was higher in S421D NPCs when compared with S421S and S421A cells post-oxidative stress induced by TBHP treatment (Fig. 6b). Moreover, we further examined whether S421 status would affect mitochondrial respiration in HD NPCs, as we had previously observed decreased oxygen consumption rate in HD NPCs when compared with corrected isogenic controls14. Under basal culture conditions, we found no differences in mitochondrial basal respiration, ATP production, or maximal respiration rate in S421A or S421D NPCs compared with S421S NPCs (Fig. 6c). However, when exposed to oxidative stress, S421D cells displayed higher ATP production and maximal respiration rate compared with S421S and S421A NPCs (Fig. 6d). These findings are consistent with the Mitotracker Red CMXRos functional mitochondria results and indicate that the S421 phosphorylation status of mHTT not only affects mitochondrial morphology but also function.

a, b NPCs were stained with functional mitochondrial marker Mitotracker Red CMXRos under basal (a) or oxidative stress (TBHP) (b) conditions; n = 6 for (a) and 8 for (b) for 2 clones per genotype with 3 BRs. Seahorse assay (adapted from Agilent website) used for measurements of mitochondrial respiration in NPCs under basal (c) and oxidative stress (TBHP) (d) conditions. Oxygen Consumption Rate (OCR) was calculated for basal respiration, ATP production, and maximal respiration; n = 3 for (C) and 5 for (D) BRs; Values shown as mean ± SEM and n.s., no significance, **p < 0.01 and ***p < 0.001 were determined by one-way ANOVA followed by a Fisher’s LSD post hoc multiple-comparison test.
Discussion
We generated isogenic HD hiPSC lines that harbor phospho-resistant and pseudo-phosphorylated residues at the S421 site of mutant HTT using TALEN- and ssODN-mediated genome editing. These isogenic hiPSC lines contain full-length HTT alleles under an endogenous promoter in the context of human physiology, and thus represent relatively ideal models in which to study the role of S421 phosphorylation in HD. We found that S421D protected HD NPCs and neurons against cell death caused by TBHP treatment and GF withdrawal. Using genome-wide gene expression analysis, we observed that S421 phosphorylation status induced a number of gene expression changes, including ones associated with mitochondrial function. Consistent with results of the transcriptional analysis, we found that the phosphorylation status of S421 did indeed affect a number of mitochondrial properties including mitochondrial numbers and morphology. Although there were no differences between S421A and S421D at baseline, mitochondrial respiration was significantly improved in S421D but not S421A under oxidative stress. Here we provide an example of leveraging genome-editing technologies in hiPSCs to study the functional significance of PTMs in HD, which may be useful for the discovery and validation of potential PTM-targeted therapeutic approaches for HD.
Single PTMs can influence protein degradation and function, and thus impact disease phenotypes2. In this study, a single amino acid change (from serine to alanine or aspartic acid) at the S421-mHTT site resulted in robust effects on HD phenotypes. Importantly, S421A and S421D manifested different effects on disease phenotypes in different assays. First, S421D, but not S421A, demonstrated protective effects under TBHP oxidative stress or the GF withdrawal assay. In addition, S421D and S421A affected mitochondrial morphology in opposite directions. For example, S421D NPCs mitochondria showed smaller surface areas, while mitochondria in S421A NPCs had bigger surface areas than that of S421S NPCs. Second, S421A and S421D behaved in a similar way in assays of neural rosette formation and basal mitochondrial respiration where neither could rescue the deficits. Thirdly, RNA-Seq data showed both S421A and S421D could affect gene expression profile. Some DEGs were specific to either S421A or S421D, while a number of DEGs were common to both groups, which is consistent with their differential and similar effects in different pathways and phenotypes. One caveat to note when interpreting the differential effects of S421A vs. S421D is that the introduction of PTM-mimicking amino acid substitutions may impact the folding state and thus influence the toxicity of mHTT, a conformationally unstable protein, which may only partly mimic the effect of endogenous PTMs.
Several studies have demonstrated that S421D protected rodent striatal neurons against mHTT-induced toxicity in vitro and in vivo, whereas S421A aggravated the toxicity9. In addition, a recent study reported that S421D-mHTT transgenic mice had less severe motor and psychiatric-like behavioral deficits than S421S-mHTT (BACHD) mice from 3 to 12 months of age, while S421A-mHTT mice only showed motor deficits at a late stage (12 months old) compared to S421S-mHTT mice24. On the other hand, both S421A and S421D were reported to be protective in mouse primary cortical and striatal neuron-based assay of mHTT toxicity in earlier studies33,34. In our study, we observed that S421D protected hiPSC-derived NPCs and neurons against cell death induced by oxidative stress and GF deprivation. In contrast, susceptibility of S421A cells to cell death was similar to that of S421S cells. Furthermore, under normal neuronal culture conditions in the presence of GFs, S421A showed more cell death, while S421D had less cell death than S421S neurons. This discrepancy among studies likely reflects the different experimental paradigms employed, such as different cell types or animal models used in the studies, different HTT expression levels, and different HTT protein length.
Clearance of mHTT is primarily mediated by the ubiquitin-proteasomal system (UPS) and autophagy-lysosomal pathway35. The UPS predominantly degrades short-lived proteins by tagging these substrates with polyubiquitin chains, while autophagy is a cellular degradative pathway for long-lived cytoplasmic proteins, protein complexes/aggregates and damaged organelles36. However, it is still unclear which of the two systems is more efficient for mHTT clearance. A recent study using transgenic S421A vs. S421D HD mice suggested that phosphorylation mitigates neurodegeneration by increasing proteasome-dependent turnover of mHTT and reducing the presence of a toxic mHTT conformer24. However, we found no obvious changes in the steady-state levels of soluble full-length mHTT levels across S421S, S421A, and S421D cells generated by a seamless genome-editing approach. When evaluating the effects of S421 phosphorylation status on the turnover of mHTT, we observed greater accumulation of mHTT in S421S NPCs when inhibiting the lysosomal pathway. This suggests the lysosomal pathway is an important pathway for mHTT degradation, and S421A and S421D might increase lysosomal-dependent turnover of mHTT. This discrepancy with the previous study24 may be due to the different species, cellular systems, or nature of the mHTT protein (full length vs. fragment) employed.
Mitochondria are highly dynamic organelles that divide, fuse, and transport. These processes not only control mitochondrial morphology and number but also regulate mitochondrial function. Strong evidence indicates that mitochondrial morphology, transport and function are affected in HD25. However, there is no report on whether phosphorylation of HTT at S421 would affect mitochondrial morphology and function. In our study, we observed increased mitochondrial mass and smaller mitochondria in S421D NPCs, which may indicate increasing mitochondrial biogenesis and fission. Increased mitochondrial biogenesis is part of cellular response to oxidative stress, as mitochondrial biogenesis can attenuate oxidative stress by increasing mitochondrial capacity to metabolize reducing equivalents, which benefits cells with better survival37,38. Moreover, mitochondrial fission is required for removal of damaged and inactive organelles by autopay, which is important for maintenance of normal mitochondrial function39. Therefore, the mitochondrial mass and morphology changes in S421D cells might be an early compensative response to HD pathogenesis. In HD hiPSC-derived neural cells, we and others24,40 have observed deficits in mitochondrial respiration. In this study, we only observed rescuing effects of S421D on mitochondrial respiration of HD NPCs under oxidative stress but not basal conditions, suggesting that loss of phosphorylation of mHTT at S421 is more likely to be consequential under conditions of cellular stress.
CHCHD2, a member of “CHCHD” domain containing protein family, is involved in mitochondrial function, and plays important roles in the biogenesis and regulation of enzymes in the mitochondrial respiratory chain41. CHCHD2 regulates mitochondrial morphology in the fat cells of flies, and loss of CHCHD2 results in the increased mitochondrial fission42. In this study, we find the CHCHD2 expression is significantly decreased in S421D cells, which may account for the lower mitochondrial surface area and volume and higher mitochondrial counts in S421 NPCs, compared with the S421A and S421S groups.
It should be noted that electron microscopy analysis of mitochondrial ultrastructure would provide a more accurate assessment of mitochondrial morphological changes associated with S421-mHTT phosphorylation status than the super-resolution microscopy employed in the present study and should be considered in future follow-up studies.
In conclusion, our study demonstrates the significant impact of S421 phosphorylation status on mitochondrial form and function, as well as susceptibility to cell death in HD hiPSC-derived neural cells under stress conditions. Our results show that PTM at S421 may modulate the toxicity of the full-length mHTT protein at least in part by affecting HD-associated mitochondrial phenotypes. These findings provide further support that targeting phosphorylation of mHTT at S421 site can be considered for further development as a therapeutic strategy for HD.
Data availability
The datasets used and/or analyzed during the current study are included in this article and its Supplementary Information files. The datasets are available from the corresponding author on reasonable request.
References
Saudou, F. & Humbert, S. The biology of huntingtin. Neuron 89, 910–926 (2016).
Ehrnhoefer, D. E., Sutton, L. & Hayden, M. R. Small changes, big impact: posttranslational modifications and function of huntingtin in Huntington disease. Neuroscientist 17, 475–492 (2011).
Ratovitski, T. et al. Post-translational modifications (PTMs), identified on endogenous huntingtin, cluster within proteolytic domains between HEAT repeats. J. Proteome Res. 16, 2692–2708 (2017).
Rangone, H. et al. The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur. J. Neurosci. 19, 273–279 (2004).
Warby, S. C. et al. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum. Mol. Genet. 14, 1569–1577 (2005).
Metzler, M. et al. Phosphorylation of huntingtin at Ser421 in YAC128 neurons is associated with protection of YAC128 neurons from NMDA-mediated excitotoxicity and is modulated by PP1 and PP2A. J. Neurosci. 30, 14318–14329 (2010).
Gauthier, L. R. et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 118, 127–138 (2004).
Warby, S. C. et al. Phosphorylation of huntingtin reduces the accumulation of its nuclear fragments. Mol. Cell. Neurosci. 40, 121–127 (2009).
Pardo, R. et al. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J. Neurosci. 26, 1635–1645 (2006).
Aiken, C. T. et al. Phosphorylation of threonine 3: implications for Huntingtin aggregation and neurotoxicity. J. Biol. Chem. 284, 29427–29436 (2009).
Jana, N. R. et al. Co-chaperone CHIP associates with expanded polyglutamine protein and promotes their degradation by proteasomes. J. Biol. Chem. 280, 11635–11640 (2005).
Steffan, J. S. et al. SUMO modification of Huntingtin and Huntington’s disease pathology. Science 304, 100–104 (2004).
Thompson, L. M. et al. IKK phosphorylates Huntingtin and targets it for degradation by the proteasome and lysosome. J. Cell Biol. 187, 1083–1099 (2009).
Xu, X. et al. Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Rep. 8, 619–633 (2017).
Li, W. et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc. Natl Acad. Sci. USA 108, 8299–8304 (2011).
Xu, X. et al. Prevention of β-amyloid induced toxicity in human iPS cell-derived neurons by inhibition of Cyclin-dependent kinases and associated cell cycle events. Stem Cell Res. 10, 213–227 (2013).
Shi, Y., Kirwan, P. & Livesey, F. J. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat. Protoc. 7, 1836–1846 (2012).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Fabregat, A. et al. Reactome pathway analysis: a high-performance in-memory approach. BMC Bioinformatics 18, 383–392 (2017).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016).
Schermelleh, L. et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 320, 1332–1336 (2008).
HD iPSC Consortium Induced pluripotent stem cells from patients with Huntington’s disease show CAG-repeat-expansion-associated phenotypes. Cell Stem Cell 11, 264–278 (2012).
Becker, B., Clapper, J., Harkins, K. R. & Olson, J. A. In situ screening assay for cell viability using a dimeric cyanine nucleic acid stain. Anal. Biochem. 221, 78–84 (1994).
Kratter, I. H. et al. Serine 421 regulates mutant huntingtin toxicity and clearance in mice. J. Clin. Invest. 126, 3585–3597 (2016).
Bossy-Wetzel, E., Petrilli, A. & Knott, A. B. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 31, 609–616 (2008).
Yano, H. et al. Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 17, 822–831 (2014).
Orr, A. L. et al. N-terminal mutant huntingtin associates with mitochondria and impairs mitochondrial trafficking. J. Neurosci. 28, 2783–2792 (2008).
Ooi, J. et al. Unbiased profiling of isogenic Huntington disease hPSC-derived CNS and peripheral cells reveals strong cell-type specificity of CAG length effects. Cell Rep. 26, 2494–2508.e7 (2019).
Feyeux, M. et al. Early transcriptional changes linked to naturally occurring Huntington’s disease mutations in neural derivatives of human embryonic stem cells. Hum. Mol. Genet. 21, 3883–3895 (2012).
Song, W. et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 17, 377–382 (2011).
Agnello, M., Morici, G. & Rinaldi, A. M. A method for measuring mitochondrial mass and activity. Cytotechnology 56, 145–149 (2008).
Pendergrass, W., Wolf, N. & Poot, M. Efficacy of MitoTracker Green and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytometry 61, 162–169 (2004).
Arbez, N. et al. Post-translational modifications clustering within proteolytic domains decrease mutant huntingtin toxicity. J. Biol. Chem. 292, 19238–19249 (2017).
Watkin, E. E. et al. Phosphorylation of mutant huntingtin at Serine 116 modulates neuronal toxicity. PLoS ONE 9, e88284 (2014).
Sarkar, S. & Rubinsztein, D. C. Huntington’s disease: degradation of mutant huntingtin by autophagy. FEBS Lett. 275, 4263–4270 (2008).
Krainc, D. Clearance of mutant proteins as a therapeutic target in neurodegenerative diseases. Arch. Neurol. 67, 388–392 (2010).
Rasbach, K. A. & Schnellmann, R. G. Signaling of mitochondrial biogenesis following oxidant injury. J. Biol. Chem. 282, 2355–2362 (2007).
Edwards, J. L. et al. Diabetes regulates mitochondrial biogenesis and fission in mouse neurons. Diabetologia 53, 160–169 (2009).
Westermann, B. Bioenergetic role of mitochondrial fusion and fission. Biochim. Biophys. Acta 1833–1838, 2012 (1817).
An, M. C. et al. Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 11, 253–263 (2012).
Aras, S. et al. Oxygen-dependent expression of cytochrome c oxidase subunit 4-2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 41, 2255–2266 (2013).
Liu, Y. et al. CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate Bax activation. Cell Death Differ. 22, 1035–1046 (2014).
Acknowledgements
We thank Weiping Yu (Biological Resources Centre, A*STAR) for reagents and members of the Pouladi lab for helpful comments. We thank the NINDS hiPSC Repository for the HD hiPSC lines (CAG180 and CAG33). The work was partly funded by a Strategic Positioning Fund for Genetic Orphan Diseases (SPF2012/005) from the Agency for Science Technology and Research (Singapore), and a Tier 1 grant R-172–000–297–112 from the Ministry of Education (Singapore) to M.A.P. C.R. is supported by grants from the CHDI Foundation and the NIH (NINDS grant R01 NS086452). This study was also supported by a grant from the National Natural Science Foundation of China (Grant Number 81901295).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by F. Strappazzon
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, X., Ng, B., Sim, B. et al. pS421 huntingtin modulates mitochondrial phenotypes and confers neuroprotection in an HD hiPSC model. Cell Death Dis 11, 809 (2020). https://doi.org/10.1038/s41419-020-02983-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41419-020-02983-z
This article is cited by
-
CHCHD2 up-regulation in Huntington disease mediates a compensatory protective response against oxidative stress
Cell Death & Disease (2024)
