
Abstract
Cardiovascular disease remains the leading cause of morbidity and mortality worldwide. Cell death is a hallmark characteristic of various cardiac diseases, including myocardial infarction and heart failure. Emerging evidences suggest that necrosis is programmed and is one of the main forms of cell death in the pathological process in cardiac diseases. However, the molecular components regulating programmed necrosis in heart remain largely unidentified. Here we report that miR-30b, Cyclophilin D (CypD) and E2F1 constitute an axis that regulates necrosis. The results show that knockdown of CypD attenuated necrosis in the cellular model and also myocardial infarction in the animal model. miR-30b suppresses the translation of CypD and thus inhibits CypD-mediated necrotic cell death in cardiomyocytes. Cardiac-specific miR-30b transgenic mice exhibit reduced necrosis and myocardial infarct size upon ischemia/reperfusion (I/R) injury. Further, we identify that E2F1 transcriptionally represses miR-30b expression. Knockdown of E2F1 in cardiomyocytes inhibits necrotic cell death, and E2F1 knockout mice show reduced necrosis and myocardial infarct size upon I/R. Our present study identifies a novel signaling pathway composed of E2F1, miR-30b and CypD that regulates myocardial necrosis. This discovery will not only provide de novo regulators in the necrotic process but will also shed new light on the effective therapy of myocardial infarction and heart failure.
Similar content being viewed by others


Main
Necrosis and apoptosis are two forms of cell death, which have key roles in the process of development, homeostasis and diseases.1 Traditionally, necrosis is regarded as a passive and unregulated cell death, while apoptosis has long been considered as the sole form of programmed cell death. However, emerging evidences suggest that a proportion of necrosis is regulated by serial signaling events in a controlled and orchestrated manner.2 Recent data have shown that programmed necrosis is a complex process and is mediated by multiple signaling pathways.3, 4, 5, 6, 7, 8 Necrosis is observed in various heart diseases, including myocardial infarction, heart failure and stroke.9, 10, 11, 12 It has been reported that death receptor pathway and mitochondrial pathway13,14 are involved in the programmed necrosis during heart disease progression. These studies show that necrotic cell death has an important role in the occurrence and development of cardiac diseases. However, the molecular components regulating programmed necrosis in the heart remain largely unidentified.
Cyclophilin D (CypD) is a prolyl isomerase that has an important role in the formation of mitochondrial permeability transition pore (mPTP).15,16 CypD, mitochondrial adenine nucleotide translocator and a voltage-dependent anion channel together forms the mPTP.16 Among the three components of the mPTP, CypD lies in the center of regulating the pore opening. And the deletion of the CypD causes marked impairment of mPTP formation.17, 18, 19 It is believed that transient opening of mPTP, caused by limited oxidative stress or short periods of ischemia, preserves the ATP/ADP ratio in the mitochondria and mainly initiates the apoptosis, while the consistent opening of mPTP usually results in ATP depletion and finally leads to necrotic cell death.20,21 As mPTP opening is a critical event in some forms of necrotic cell death, the function of CypD is also important in the necrotic cell death pathway. Previous reports have shown that the mPTP in the inner mitochondrial membrane is involved in the mitochondrial pathway and CypD mediates the programmed necrosis.7,8 CypD has recently been shown to be able to regulate hydrogen peroxide (H2O2)-induced necrotic cell death.22,23 However, it is not yet clear whether CypD is a target of microRNAs (miRNAs), and the molecular regulation of CypD in the necrotic machinery remains to be elucidated. Addressing these questions will be of great importance to decipher and characterize the molecular pathway of necrosis.
miRNAs are a class of short single-stranded non-coding endogenous RNAs and act as negative regulators of gene expression by inhibiting mRNA translation or promoting mRNA degradation.24,25 Although the function of miRNAs has been widely studied in apoptosis, development, differentiation and proliferation, few works have focused on the function of miRNAs in the necrotic pathway. It has been reported that miR-155 targets to RIP1 and prevents cardiomyocyte progenitor cells’ necrosis.26 And miR-214 protects heart from ischemia/reperfusion (I/R) injury and necrotic cell death by targeting to NCX1 and inhibiting Ca2+ overload.27 The study of miRNA function in cardiomyocyte necrosis will shed new light on the necrotic cell death pathway and discover de novo therapeutic targets for heart failure.
E2F1 lies downstream of retinoblastoma protein, promotes apoptosis and suppresses cell proliferation.28,29 E2F1 is pivotal for cardiac function and mainly affect the metabolism of cardiomyocytes. E2F1 knockout mice have lower blood glucose levels and improved glucose oxidation and shows a variety of age-related defects.30 E2F1 mutation induces congestive heart failure.31 These studies further substantiate the notion that E2F1 might have an important role in the pathological development of heart disease. So far, few studies have focused on the E2F1 function in cell necrosis. As apoptosis and necrosis both exist in heart failure, it is likely that E2F1 participates in necrosis signaling pathway. It is of great importance to study and unveil the function of E2F1 in necrosis.
Our present study unveils that CypD is involved in the regulation of myocardial necrosis. Knockdown of CypD inhibits necrotic cell death in vivo and in vitro. In searching for the mechanism by which CypD is upregulated under necrotic condition, we identify that miR-30b participates in the suppressing of CypD translation. miR-30b inhibits necrotic program through targeting CypD. We further find that E2F1 transcriptionally represses miR-30b expression. E2F1 initiates necrotic program through downregulating miR-30b. Our results reveal a novel myocardial necrotic-regulating model, which is composed of E2F1, miR-30b and CypD. Modulation of their levels will shed new light on the necrotic cell death pathway and may represent a novel approach for interventional treatment of myocardial infarction and heart failure.
Results
CypD regulates H2O2-induced necrotic cell death in cardiomyocytes
It is well known that H2O2 induces necrotic cell death in various cell types.32, 33, 34 CypD, a protein considered to be involved in mPT, has recently been shown to be able to regulate H2O2 -induced necrotic cell death.22 However, the underlying molecular mechanism of CypD in necrosis remains to be elucidated. We tested whether CypD is related to the occurrence of necrosis induced by H2O2 in cardiomyocytes. We observed that H2O2 treatment led to a time-dependent increase in CypD expression (Figure 1a). Knockdown of CypD attenuated CypD levels and necrotic cell death (Figure 1b) induced by H2O2. These data suggest that CypD is involved in H2O2-induced necrotic cell death and has a functional role in caridomyocytes.

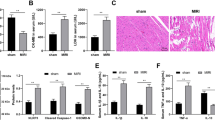
CypD is involved in H2O2-induced necrotic cell death in caridomyocytes. (a) CypD levels are upregulated upon H2O2 treatment. Cardiomyocytes were treated with 750 μM H2O2 at the indicated times. CypD levels were analyzed by immunoblot. (b) Knockdown of CypD reduces necrotic responses induced by H2O2. Cardiomyocytes were infected with an adenoviral CypD-siRNA or its scramble form (CypD-sc). Twenty-four hours after infection, cells were treated with H2O2. The expression levels of CypD were analyzed by immunoblot (upper panel) and necrotic cell death was assessed by PI exclusion assay (low panel); *P<0.05 versus H2O2 alone. (c) The levels of CypD are increased in myocardial I/R. Mice were induced to undergo cardiac I/R at the indicated times as described in Materials and Methods. CypD levels were analyzed by immunoblot. (d) Knockdown of CypD attenuates myocardial necrosis upon I/R. Mice were subjected to I/R as described in Materials and Methods. The percentage of cells with myosin antibody infiltration was shown. n=6, *P<0.05 versus I/R alone. (e) Knockdown of CypD inhibits myocardial infarction. Mice were treated as described in panel (d). Infarct size were shown. n=6. *P<0.05 versus I/R alone. (f) Knockdown of CypD preserves cardiac function upon I/R. Transthoracic echocardiographic analysis was performed after 60-min ischemia followed by 1-week reperfusion. Fractional shortening (FS) of LV diameter is shown. *P<0.05 versus I/R alone. GAPDH, glyceraldehyde 3-phosphate dehydrogenase
To understand the pathophysiological role of CypD, we tested whether CypD is involved in the pathogenesis of myocardial infarction in the animal model. Our results showed that CypD was elevated in response to I/R injury (Figure 1c). Myosin antibody injection technique was used to assess myocardial necrosis. I/R caused a massive increase in necrotic myosin-positive cells (Figure 1d). Knockdown of CypD by siRNA delivery showed a decrease in necrotic myosin-positive cells compared with the control group upon I/R (Figure 1d). Thus it indicates that knockdown of CypD results in a reduction in the myocardial necrosis. The myocardial infarct size was also reduced in the CypD knockdown group (Figure 1e), and the cardiac function was ameliorated by knockdown of CypD (Figure 1f). Taken together, it appears that CypD has a significant role in the regulation of myocardial necrosis and myocardial infarction.
miR-30b participates in the regulation of CypD expression
miRNAs are a class of small non-coding RNAs and act as negative regulators of gene expression. To explore the underlying mechanism by which CypD is upregulated upon H2O2 and I/R treatment, we tested whether CypD is regulated by miRNAs. We first screened some cardiac-associated miRNAs, which had been reported in the past several years by quantitative reverse transcription-PCR (qRT-PCR). Among these miRNAs, miR-30b levels were significantly downregulated upon H2O2 (Figure 2a), and other miRNAs remain unchanged (data not shown). We also analyzed the 3′UTR of CypD using the RNAhybrid program and observed that CypD was a potential target of miR-30b (Figure 2b), which promoted us to focus on the function of miR-30b. To identify the function of miR-30b, several approaches were used to explore whether CypD expression is regulated by miR-30b. We first tested whether overexpression or knockdown of miR-30b altered the expression of endogenous CypD. Our results showed that enforced expression of miR-30b resulted in a reduction of endogenous CypD (Figure 2c). Concomitantly, knockdown of endogenous miR-30b induced an increase in CypD expression (Figure 2d). Enforced expression of miR-30b also led to a reduction of CypD upon H2O2 treatment (Figure 2e). These results indicate that miR-30b can specifically regulate CypD.

miR-30b participates in the regulation of CypD expression. (a) qRT-PCR analysis of miR-30b. Cardiomyocytes were treated with H2O2 at the indicated times, and the expression of miR-30b was analyzed. *P<0.05 versus control. (b) Putative miR-30b sites in the 3′UTR (untranslated region) region of CypD. This putative miR-30b site in the CypD 3′UTR region was analyzed by the RNAhybrid program. (c) miR-30b suppresses the expression of CypD in cardiomyocytes. Cardiomyocytes were infected with adenoviral miR-30b or β-gal. CypD expression was analyzed by immunoblot. (d) Knockdown of miR-30b induces the increase of CypD expression levels. Cardiomyocytes were transfected with antagomir miR-30b (anta-30b) or its negative control (anta-NC). Forty-eight hours after transfection, CypD expression levels were analyzed by immunoblot. (e) miR-30b reduces the CypD expression levels induced by H2O2. Cardiomyocytes were infected with adenoviral miR-30b or β-gal. Twenty-four hours after infection, cells were treated with H2O2. The expression levels of CypD were analyzed by immunoblot. (f) CypD WT 3′UTR and a mutated 3′UTR in the miR-30b-binding site are shown. (g) Luciferase assay. HEK293 cells were infected with adenoviral miR-30b or β-gal and then transfected with the luciferase constructs of the WT CypD-3′UTR (CypD-3′UTR-wt) or a mutated CypD-3′UTR (CypD-3′UTR-mut). The luciferase activity was analyzed. *P<0.05. (h) miR-30b can suppress the expression of CypD with WT but not mutated UTR. Cardiomyocytes were co-infected with the adenoviral miR-30b or β-gal, along with CypD with WT UTR (CypD-3′UTR-wt) or the mutated UTR (CypD-3′UTR-mut). CypD levels were detected by immunoblot. (i) miR-30b suppresses the expression of CypD in the animal model. CypD expression was analyzed by immunoblot from WT and miR-30b transgenic mice of lines 1, 2 and 3. GAPDH, glyceraldehyde 3-phosphate dehydrogenase
We employed the luciferase assay system to test whether miR-30b influenced the translation of CypD. As shown in Figure 3g, the luciferase reporter assay revealed that the wild-type (WT) 3′UTR of CypD exhibited a low translation level in the presence of miR-30b, whereas the mutated 3′UTR (Figure 2f) did not show a significant response to miR-30b. Our data further showed that miR-30b reduced the expression of CypD with WT 3′UTR (Figure 2h, upper panel) but had no effect on the mutated 3′UTR (Figure 2h, low panel). To detect the role of miR-30b in the regulation of CypD in animal model, we generated transgenic mice with cardiac-specific overexpression of miR-30b (Supplementary Figure S1A). Five lines of miR-30b transgenic mice represented a high level of miR-30b in the heart (Supplementary Figure S1B). Our results showed that CypD expression level was downregulated in miR-30b transgenic mice (Figure 2i). Taken together, it suggests that CypD is a specific target of miR-30b.

miR-30b is able to regulate necrosis in vitro and in vivo. (a and b) miR-30b reduces necrotic responses induced by H2O2. Cardiomyocytes were transfected with mimic miR-30b (mimic-30b) or mimic control (mimic-NC). Twenty-four hours after transfection, cells were treated with H2O2. Necrosis was assessed by EM (a) and PI exclusion (b). *P<0.05 versus H2O2 alone. (c) miR-30b levels during myocardial I/R. Mice were induced to undergo cardiac I/R. AAR and remote area were prepared at the indicated times for qRT-PCR analysis of miR-30b levels. n=6, *P<0.05 versus 0 min or sham. (d and e). miR-30b attenuates myocardial necrosis upon I/R injury. miR-30b transgenic mice (Tg) and WT mice were subjected to I/R as described in Materials and Methods. (d) Immunohistochemistry for myosin antibody incorporation into the heart (green); red is a wheat germ stain for cell membranes; nuclei are shown in blue. The percentage of cells with myosin antibody infiltration is shown (e). n=6, *P<0.05 versus WT+I/R. (f) miR-30b inhibits myocardial infarction. miR-30b transgenic mice (Tg) and WT mice were treated as described in panel (e). Infarct size was shown. n=6. *P<0.05 versus WT+I/R. (g) miR-30b preserves cardiac function upon I/R. Transthoracic echocardiographic analysis was performed after 60-min ischemia followed by 1-week reperfusion. Fractional shortening (FS) of LV diameter was shown. *P<0.05 versus WT+I/R
miR-30b is able to inhibit necrotic cell death in vitro and in vivo
Next, we attempted to understand the function of miR-30b in necrotic machinery. Necrosis is morphologically characterized by increasing of cell volume, dilation of organelles, rupture of the plasma membrane and subsequent loss of intracellular contents. Cardiomyocytes upon exposure to H2O2 (750 μM) displayed obvious necrotic morphological characterization. However, compared with the mimic negative control (mimic-NC), enforced expression of miR-30b by the mimic (mimic-30b) group attenuated the necrotic morphological hallmarks, which were confirmed by electron microscopy (Figure 3a). miR-30b also attenuated necrotic cell death induced by H2O2 (Figure 3b). These data suggest that miR-30b has a fundamental role in H2O2-induced cardiomyocyte necrosis.
We further detected the role of miR-30b in the pathogenesis of myocardial infarction in the animal model. We observed that miR-30b level was reduced in response to I/R injury (Figure 3c). To better understand the function of miR-30b in the heart, we generated transgenic mice with cardiac-specific overexpression of miR-30b. Analysis of adult mice generated from five stable independent transgenic mice founder lines showed that their miR-30b expression was about three to six times as high as that of the WT hearts (Supplementary Figure S1B). The miR-30b transgenic mice generated from Line#1 were used for this study (Supplementary Figure S1C). All five transgenic mice founder lines developed normally to adulthood, without significant alterations in terms of phenotype and cardiac function (Supplementary Figure S1D) under physiological condition. Next, we used myosin antibody injection technique to assess myocardial necrosis. miR-30b transgenic mice showed less myosin antibody incorporation than the WT group upon I/R, which indicated a reduction in myocardial necrosis (Figures 3d and e). Myocardial infarct size also reduced in miR-30b transgenic mice, compared with WT group upon I/R (Figure 3f). The cardiac function was ameliorated in miR-30b transgenic mice group after I/R (Figure 3g). Thus, our results strongly suggest a possible contribution of miR-30b in inhibiting necrosis in the I/R-injured heart.
In addition to Line#1, we also conducted experiments using transgenic mice generated from Line#4, which have the highest miR-30b expression level among all five lines. In comparison with transgenic mice Line#1, the hearts of transgenic mice Line#4 showed an even more reduced CypD expression level (Supplementary Figure S2A) and a smaller infarct size (Supplementary Figure S2B), indicating that the functional effect of miR-30b is dependent upon the level of transgene expression.
miR-30b regulates necrosis through targeting CypD
We then explored how miR-30b exerts its effect on necrotic program. As miR-30b is able to suppress CypD expression, we thus tested whether CypD is a mediator of miR-30b. Although miR-30b could inhibit CypD expression and necrotic cell death, this inhibitory effect was significantly abolished by CypD with mutated 3′UTR but not the CypD with WT 3′UTR (Figures 4a and b). To confirm the relationship between miR-30b and CypD in necrosis program machinery, we employed the target protector (TP) technology in which a TP is able to disrupt the specific interaction of miRNA–mRNA pairs. To this end, we produced CypD TP and observed that the inhibitory effect of miR-30b on CypD expression and necrotic cell death was reduced in the presence of CypD TP (Figures 4c and d). These data suggest that miR-30b specifically targets CypD in the signal transduction cascades of necrotic program.

miR-30b regulates necrosis through targeting CypD. (a and b) miR-30b can attenuate CypD levels and necrotic responses in the presence of CypD with WT untranslated region (UTR) (CypD-3′UTR-wt) but not its mutated form (CypD-3′UTR-mut). Cardiomyocytes were infected with the adenoviral miR-30b, along with the constructs of CypD-3′UTR-wt or CypD-3′UTR-mut. Twenty-four hours after infection, cells were treated with H2O2.The expression levels of CypD were detected by immunoblot (a). Necrotic cell death was assessed by PI exclusion assay (b). *P<0.05. (c and d). CypD TP reduces the inhibitory effect of miR-30b on CypD expression and necrosis. Cardiomyocytes were infected with adenoviral miR-30b and transfected with the TP (CypD-TP miR-30b) or the control (CypD-TP control). CypD expression was analyzed by immunoblot (c). Necrotic cell death was assessed by PI exclusion assay (d). *P<0.05. GAPDH, glyceraldehyde 3-phosphate dehydrogenase
miR-30b is negatively regulated by E2F1 in transcriptional level
We asked how miR-30b expression is regulated under the pathological condition. It has recently been reported that NF-κB positively regulates miR-30b expression in Ang II-induced cardiomyocyte apoptosis, and Bcl-2 is a direct target of miR-30b.35 Thus we analyzed the expression levels of NF-κB and BCL-2 to explore whether they are also involved in the necrotic pathway induced by H2O2. The results showed that there is no detectable change in the expression levels of NF-κB and BCL-2 in response to H2O2 treatment (Supplementary Figure S3A), suggesting that NF-κB and BCL-2 are not involved in H2O2-induced necrotic pathway.
We then analyzed the promoter region of mouse miR-30b and observed that there was a potential binding site of E2F1 (Figure 5a). Enforced expression of E2F1 led to a decrease of miR-30b precursor (pre-miR-30b) and miR-30b levels (Supplementary Figure S3B and Figure 5b), whereas E2F1 knockout mice exhibited an increased expression levels of pre-miR-30b and miR-30b (Supplementary Figure S3C and Figure 5c), suggesting that miR-30b may be a potential transcriptional target of E2F1. Thus we tested whether E2F1 can bind to the promoter region of miR-30b. The ChIP assay revealed that E2F1 bound to the miR-30b promoter under the physiological condition. H2O2 treatment enhanced the association of E2F1 with the miR-30b promoter (Figure 5d). We further tested whether E2F1 could influence miR-30b promoter activity. WT miR-30b promoter (wt) represented a low activity in the presence of E2F1. And mutations in the E2F1-binding site could abolish the inhibitory effect of E2F1 on miR-30b promoter activity (Figure 5e). These data suggest that E2F1 can negatively regulate the expression of miR-30b. H2O2 induced a reduction in miR-30b promoter activity in cardiomyocyte (Figure 5f). Concomitantly, knockdown of E2F1 could attenuate the decrease of miR-30b promoter activity induced by H2O2 (Figure 5f). These data indicate that miR-30b can be transcriptionally repressed by E2F1.

miR-30b is a transcriptional target of E2F1. (a) Mouse miR-30b promoter region contains a potential E2F1-binding site. (b) E2F1 inhibits miR-30b expression. Cardiomyocytes were infected with adenoviral β-gal or E2F1. miR-30b levels were analyzed by qRT-PCR (upper panel). *P<0.05 versus control. E2F1 expression was analyzed by immunoblot (low panel). (c) miR-30b levels in E2F1-deficient mice. E2F1−/−, E2F1 knockout mice, WT, wild-type littermates. n=5. *P<0.05 versus WT. (d) ChIP analysis of E2F1 binding to the promoter of miR-30b. (e) E2F1 suppresses miR-30b promoter activity. Cardiomyocytes were treated with the adenoviral β-gal or E2F1, the constructs of the empty vector (pGL-4.17), the WT promoter (wt) or the promoter with mutations in the binding site (mutant), respectively. Luciferase activity was assayed. *P<0.05. (f) Knockdown of E2F1 attenuates the decrease of miR-30b promoter activity induced by H2O2. Cardiomyocytes were treated with the adenoviral E2F1-siRNA or E2F1-sc, the constructs of the empty vector (pGL-4.17), the WT promoter (wt) and then were treated with H2O2. Luciferase activity was assayed. *P<0.05. GAPDH, glyceraldehyde 3-phosphate dehydrogenase
Our data have shown that CypD acts as a specific downstream target of miR-30b, and miR-30b transcription, in turn, is regulated by E2F1. Thus we explored whether there is a direct interaction between E2F1 and CypD. We cloned the promoter region of mouse CypD and constructed the luciferase reporter vector. Luciferase activity analysis showed that E2F1 did not influence the CypD promoter activity (Supplementary Figure S3D), suggesting that CypD can not be directly regulated by E2F1 at the transcriptional level. Thus our results indicate that there is no direct interaction between E2F1 and CypD.
E2F1 regulates necrosis through miR-30b and CypD
We investigated the role of E2F1 in cardiomyocyte necrosis and myocardial infarction. H2O2 induced an elevation in E2F1 expression level (Figure 6a). Knockdown of E2F1 was able to attenuate necrotic cell death upon H2O2 treatment in cardiomyocytes (Figure 6b). I/R also led to an increase in E2F1 expression level in response to I/R injury. Furthermore, E2F1 knockout mice demonstrated reduced necrotic cell death revealed by myosin antibody incorporation (Figure 6d) and myocardial infarction size (Figure 6e) upon I/R. These data indicate that E2F1 participates in mediating the signal for necrotic cell death in the heart.

E2F1 promotes necrosis through miR-30b and CypD. (a) E2F1 levels are increased in cardiomyocytes exposed to H2O2. Cardiomyocytes were exposed to H2O2. Cells were harvested at the indicated times for the analysis of E2F1 levels by immunoblot. (b) Knockdown of E2F1 reduces necrotic cell death induced by H2O2. Cardiomyocytes were infected with adenoviral E2F1-siRNA or E2F1-sc. Twenty-four hours after infection, cells were treated with H2O2. PI exclusion was analyzed. *P<0.05 versus H2O2 alone. (c) The levels of E2F1 are increased in myocardial I/R. Mice were induced to undergo cardiac I/R at the indicated times as described in Materials and Methods. E2F1 levels were analyzed by immunoblot. (d and e) E2F1 knockout mice attenuates myocyte necrosis and myocardial infarction upon I/R. WT and E2F1 knockout mice were subjected to I/R as described in Materials and Methods. Myocyte necrosis (d) and myocardial infarction (e) were analyzed. *P<0.05 versus WT+I/R. (f and g) CypD TP attenuates the inhibitory effect of E2F1 knockdown on CypD expression and necrotic responses induced by H2O2. Cardiomyocytes were infected with adenoviral E2F1-siRNA or E2F1-sc, transfected with the TP (CypD-TP miR-30b) or the control (CypD-TP control) and then exposed to H2O2. CypD expression (f) was analyzed by immunoblot. Necrosis was assessed by PI exclusion assay (g). *P<0.05. GAPDH, glyceraldehyde 3-phosphate dehydrogenase
We further explored whether miR-30b and CypD could act as the downstream targets of E2F1 in necrotic signal pathway. We observed that the TP of CypD attenuated the inhibitory effect of E2F1 knockdown on CypD expression (Figure 6f) and necrotic cell death (Figure 6g). Thus it appears that E2F1 targets miR-30b/CypD axis in the necrotic cascades.
Besides cardiomyocytes, many different types of cells exist in the heart, such as macrophages, monocytes, neutrophils and so on, which are also important for a properly functioning heart. It has been reported that cells other than cardiomycoytes also contribute to I/R injury.36 In consideration of the extra-cardiomyocyte effects, we examined whether other cells in turn have an important role in I/R. Our results revealed that the anti-apoptotic effect of E2F1 KO mice is not dependent on endothelium effect (Supplementary Figure S4A and Supplementary Figure S4B). Also, E2F1 KO mice do not affect miR-30b and CypD expression levels in macrophages (Supplementary Figure S5A and Supplementary Figure S5B), hence excluding the effect of these two kinds of cells in our animal models. Our results do not completely rule out the possibility of the effect of other cells, such as monocytes and neutrophils. And these cells might be promising targets for ameliorating I/R injury. In the future, we intend to carry out more rigorous studies on the effects of other inflammatory cells on cardiac I/R injury. Future studies may clarify the most promising molecules and cell types to be investigated as targets for cardiovascular diseases, providing valuable suggestions for an effective all-round treatment and a better understanding of the molecular mechanisms in a wider setting.
Discussion
Heart failure is one of the leading causes of hospitalization and death worldwide. Cardiac myocyte necrosis is an important component of myocardial infarction and heart failure. Antagonizing the myocardial necrosis is considered to be a therapeutic target for treating heart failure. So it is necessary to elucidate the molecular mechanisms of myocardial necrosis and discover potential therapeutic targets for inhibiting myocardial necrosis and the consequent heart failure. Our present work demonstrated that knockdown of CypD attenuates necrotic cell death in cardiomyocytes and reduces myocardial infarction in the animal model. miR-30b could inhibit myocardial necrosis through targeting CypD. miR-30b transgenic mice exhibit attenuated necrotic cell death and myocardial infarction size. We further identified that knockdown of E2F1 inhibits myocardial necrosis through negatively regulating miR-30b at the transcriptional level. Our study reveals a novel molecular mechanism of myocardial necrosis and provides the therapeutic targets for treating myocardial infarction and heart failure.
Necrosis is characterized by marked swelling of cell, rupture of the plasma membrane and severe inflammation followed by the spill of intracellular contents. Traditionally, necrosis is regarded as a form of accidental and unregulated cell death, while apoptosis has long been considered as the sole form of programmed cell death. However, it is clear now that a portion of necrosis is also programmed and can be actively regulated.2 Recent reports have described that two signaling pathways, including death receptor pathway3, 4, 5, 6 and mitochondrial pathway,7,8 could mediate programmed necrosis. However, there is no doubt that the study of necrosis signaling is just beginning. The molecular mechanisms of necrosis need to be further elucidated. Our present work reveals a novel programmed necrotic pathway composed of E2F1, miR-30b and CypD. The results not only expand our knowledge of the molecular mechanism of programmed necrosis but also provide new insights for understanding the programmed necrosis pathogenesis of myocardial necrosis.
mPTP has an important role in necrotic regulation. CypD knockout mice is resistant to I/R and represents reduced infarct size. Knockout of CypD mainly preserves the cardiac function through inhibiting of myocardial necrosis.22,23 The above evidences showed that CypD is an important effector in regulating necrosis. However, the underlying upstream regulator of CypD in necrosis remains unknown. Our research discovers a signaling pathway composed of E2F1, miR-30b and CypD. E2F1 and miR-30b are the upstream regulators of CypD. Thus the discovering of the upstream signaling pathway of CypD will shed new light on the regulation of programmed necrosis and will unveil novel therapeutic targets for myocardial infarction at the same time.
miRNAs participates in various biological and pathological processes, such as apoptosis, development and proliferation. Many studies have focused on the inhibitory effect of miR-30b in apoptosis in various cancerous diseases, such as lung cancer, glioma and breast cancer. miR-30b is upregulated in many cancer cells that is resistant to apoptosis. For example, miR-30b expression level is upregulated in the TRAIL-resistant cancer cells and inhibits apoptosis by targeting caspase-3.37 miR-30b targets p53 and inhibits mitochondrial fission during apoptosis.38 However, few studies reported the function of miR-30b in necrosis, which is another important regulated form of cell death in I/R injury. Our results identified that miR-30b inhibits necrotic program by inhibiting CypD expression in cardiomyocytes. And miR-30b cardiac-specific transgenic mice represent reduced myocardial necrosis and infarction size upon I/R injury. Our study has provided important evidences about the function of microRNAs in the programmed necrosis pathway, a step toward better understanding the miRNA based therapy for I/R injury and heart failure.
Necrosis and apoptosis both exist during I/R.39 In the early stage of ischemia, apoptosis is the major form of cell death, whereas in the later period of ischemia necrosis causes the majority of cardiomyocyte death. It is reported that E2F1 expression is increased upon I/R injury, and I/R injury is attenuated in E2F1 null hearts.40 The mechanism of E2F1 in inducing apoptosis has been widely studied.41 However, few studies have been focused on the E2F1 function in cell necrosis. In the present work, we investigated the role of E2F1 in necrosis upon myocardial I/R injury. E2F1 knockout mice demonstrated reduced necrotic cell death and myocardial infarction size upon I/R injury. E2F1 participates in the necrotic pathway by regulating the expression of miR-30b. Modulation of their levels may provide a new approach for tackling necrotic cell death and myocardial infarction.
Materials and Methods
Cardiomyocyte culture and treatment
Cardiomyocytes were isolated from 1 to 2 days old mice as described by us.42 Briefly, after dissection, the hearts were washed and minced in HEPES-buffered saline solution. Tissues were then dispersed in a series of incubations at 37 °C in HEPES-buffered saline solution containing 1.2 mg/ml pancreatin and 0.14 mg/ml collagenase (Worthington, Lakewood, NJ, USA). After centrifugation, cells were re-suspended in Dulbecco’s modified Eagle medium/F-12 (GIBCO, Grand Island, NY, USA) containing 5% heat-inactivated horse serum, 0.1 mM ascorbate, insulin-transferring-sodium selenite media supplement (Sigma, St Louis, MO, USA), 100 U/ml penicillin, 100 μg/ml streptomycin and 0.1 mM bromodeoxyuridine. The dissociated cells were pre-plated at 37 °C for 1 h. The cells were then diluted to 1 × 106 cells/ml and plated in 10 μg/ml laminin-coated different culture dishes according to the specific experimental requirements. Cells were treated with 750 μM H2O2 except as otherwise indicated elsewhere.
Generation of cardiac-specific miR-30b transgenic mice
For creating miR-30b transgenic mice, a 471-bp DNA fragment containing murine miR-30b was cloned to the vector, pαMHC-clone26 (kindly provided by Dr Zhong Zhou Yang), under the control of the α-myosin heavy chain (α-MHC) promoter. The primers used to generate miR-30b transgenic mice include forward primer 5′-TGAAAGAGAGAACGATAAATGTT-3′ and reverse primer: 5′-ACTTCTGAATCAAAATATTGGTA-3′. Microinjection was performed following the standard protocols.
Generation of E2F1 knockout mice
E2F1 knockout (KO) mice were purchased from Mutant Mouse Regional Resource Center (University of California, Davis, CA, USA). E2F1+/− mice were interbred to give knockout mice (E2F1−/−), which were used for further studies. Mice were genotyped by multiplex PCR (primers and conditions are available from Mutant Mouse Regional Resource Center). All experiments were performed on E2F1−/− mice and their WT littermates (E2F1+/+). The animal experiments were performed according to the protocols approved by the Institute Animal Care Committee and were approved by government authorities.
Cell death assays
Necrotic cell death was assessed by propidium iodide (PI) exclusion.
Isolation of macrophages
Mice were killed on day 7 after myocardial infarction. The heart was dissected out and perfused with cold phosphate-buffered saline. Then the heart was dissociated into a single-cell suspension as previously described.43 Briefly, the heart was minced and digested with a cocktail composed of collagenase and DNase I for 30 min at 37 °C with rotation. BSA was added to the digested samples. Cells were centrifuged and resuspended with RMPI-1640 cell culture medium, supplemented with 10% FBS and incubated in a plastic dish at 37 °C. Two hours later, adherent cells were collected and stained with F4/80-PE and cd11b-PE-cy5 for FACS analysis. F4/80 and CD11b double-positive macrophages were sorted and collected. RNA was extracted from the sorted macrophage using the microElute total RNA kit (Omega, Norcross, GA, USA).
Transfection of antagomir
miR-30b antagomir and the antagomir negative control (antagomir-NC) were purchased from GenePharma Co. Ltd (Shanghai, China). The antagomir sequence is 5′-GACGUAAACAUCCACAUCCCAG-3′. All the bases were 2′-OMe modified, and the 3′-end was conjugated to cholesterol. Chemically modified oligonucleotides 5′-CAGUACUUUUGUGUAGUACAA-3′ were used as a negative control (antagomir-NC). Cells were transfected with the antagomir or antagomir-NC at 50 nM. The transfection was performed using Lipofectamine 2000 (Invitrogen, GrandIsland, NY, USA) according to the manufacturer’s instruction.
Target protector preparation and transfection
Target protector was designed and named as described by others and us.44 In brief, CypD-TPmiR-30b sequence is 5′-CTGGGTGAATCAAGACATGGATGCCT-3′ and CypD-TPcontrol sequence is 5′-TGACAAATGAGACTCTCTCCTCTCC-3′. They were synthesized by Gene Tools and transfected into the cells using the Endo-Porter kit (Gene Tools, Philomath, OR, USA) according to the kit’s instructions.
Preparations of the luciferase construct of CypD 3′UTR and luciferase activity assay
CypD 3′UTR was amplified by PCR. The forward primer was 5′-CAAGGTGCTAGGACAGCAGCA-3′; the reverse primer was 5′-CCCTGAGTGATGTCTCAGAAG-3′. To produce mutated 3′UTR, the mutations were generated using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). The constructs were sequence verified. WT and mutated 3′UTRs were subcloned into the pGL3 vector (Promega, Madison, WI, USA) immediately downstream of the stop codon of the luciferase gene.
Luciferase activity assay was performed using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Cells were co-transfected with the plasmid constructs of 150 ng/well of pGL3-CypD-3′UTR or pGL3-CypD-3′UTR-mut using Lipofectamine 2000 (Invitrogen) and then were infected with adenovirus miR-30b or β-gal at an MOI of 80. At 48 h after infection, luciferase activity was measured.
Adenoviral constructions and infection
Mouse CypD cDNA was amplified by RT-PCR from total RNA of mouse heart and then cloned into pcDNA3.1. The adenoviruses harboring CypD-3′UTR-wt were constructed using the Adeno-X expression system (Clontech, Otsu, Japan). CypD 3′UTR mutants were generated using the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene). Mouse E2F1 cDNA was from Origene (Rockville, MD, USA). The adenoviruses harboring E2F1 was constructed using the Adeno-X expression system (Clontech). To construct adenovirus encoding miR-30b, the mature miR-30b sequence was cloned into the adenovirus system according to the manufacturer’s instructions.
Constructions of adenoviruses harboring mouse CypD RNAi and E2F1-siRNA
The mouse CypD RNAi target sequence is 5′-GTCTTCCTCCTTGCAATAATT-3′. A non-related, scrambled RNAi without any other match in the mouse genomic sequence was used as a control (5′-GTTTCAACTTCCTGTACTACT-3′). E2F1 siRNA sequence is 5′-ATCTGACCACCAAACGCTT-3′; the scramble E2F1 siRNA sequence is 5′-CACTCACGACTACGATCTA-3′. The adenoviruses harboring these RNAi constructs were generated using the pSilencer adeno 1.0-CMV System (Ambion, GrandIsland, NY, USA) according to the Kit’s instructions.
Constructions of mouse miR-30b promoter and its mutant
The miR-30b promoter was amplified from mouse genome using PCR. The forward primer was 5′-GCTTCTAATGTTGGGAGCTGA-3′. The reverse primer was 5′-TGGGAGCCAAGCAATAACCTA-3′. The promoter fragment was finally cloned into the vector pGL4.17 (Promega). The introduction of mutations in the putative E2F1-binding site was performed with the QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene) using the WT vector as a template. The construct was sequenced to check that only the desired mutations had been introduced.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed as described by us.44 In brief, cells were washed with PBS and incubated for 10 min with 1% formaldehyde at room temperature. The cross-linking was quenched with 0.1 M glycine for 5 min. Cells were washed twice with PBS and lysed for 1 h at 4 °C in a lysis buffer. The cell lysates were sonicated into chromatin fragments with an average length of 500–800 bp as assessed by agarose gel electrophoresis. The samples were precleared with Protein-A agarose (Roche, Hamburg, Germany) for 1 h at 4 °C on a rocking platform, and 5 μg specific antibodies were added and rocked for overnight at 4 °C. Immunoprecipitates were captured with 10% (vol/vol) Protein-A agarose for 4 h. Before use, Protein-A agarose was blocked twice at 4 °C with salmon sperm DNA (2 μg/ml) overnight. DNA fragments were purified with a QIAquick Spin Kit (Qiagen). The purified DNA was used as a template and amplified with the following primer sets: For the analysis of E2F1-binding to the promoter region of miR-30b, the oligonucleotides were as follows: forward: 5′-GCTTCTAATGTTGGGAGCTGA-3′; and reverse: 5′-GTAGACATGAATAACCTACAGAG-3′.
Immunoblot
Immunoblot was performed as we described.45 The anti-CypD antibody was from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA). The anti-E2F1 antibody, anti-Bcl-2 antibody and the anti-GAPDH antibody were from Abcam (GrandIsland, NY, USA). Anti-PECAM-1 (CD31) antibody was from Millipore (Billerica, MA, USA), and anti-p65 antibody was from Cell Signaling Technology (Danvers, MA, USA). The horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology, Inc.
Quantitative reverse transcription-PCR
Stem-loop qRT-PCR for mature miR-30b was performed as described46 on a CFX96 Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Total RNA was extracted using the Trizol reagent. After DNAse I (Takara, Otsu, Japan) treatment, RNA was reverse transcribed with reverse transcriptase (ReverTra Ace, Toyobo, Japan). The levels of miR-30b analyzed by qRT-PCR were normalized to that of U6. U6 primers were forward: 5′-GCTTCGGCAGCACATATACTAA-3′; and reverse: 5′-AACGCTTCACGAATTTGCGT-3′.
Northern blot analysis
Northern blot was performed as described.47 In brief, the samples were run on a 15% polyacrylamide-urea gel, transferred to positively charged nylon membranes (Millipore) followed by cross-linking through UV irradiation. The membranes were subjected to hybridization with 100 pmol 3′-digoxigenin (DIG)-labeled probes for miR-30b overnight at 42 °C. miR-30b probes were labeled with DIG using a 3′-End DIG Labeling Kit (Roche). The detection was performed using a DIG luminescent detection kit (MyLab) according to the manufacturer’s instructions. The probe sequence for miR-30b was 5′-GACGTAAACATCCACATCCCAG-3′. DIG-labeled U6 probe was used as an internal control, and its sequence was 5′-TGGAACGCTTCACGAATTTG-3′.
Transmission electron microscopy
Conventional electron microscopy was performed as described previously.48 In brief, cells were fixed with 2.5% glutaraldehyde and then postfixed with 1% osmium tetraoxide, dehydrated in a graded series of ethanol concentrations and embedded in Embed812 resin. The ultrathin sections were mounted on copper grids and then double-stained with uranyl acetate and lead citrate. The samples were examined and photographed with a FEI Tecnai spirit transmission electron microscope (Hillsboro, OR, USA).
I/R, preparations of area-at-risk (ARR) and histology
Male adult C57BL/6 mice (8-weeks old) were obtained from the Institute of Laboratory Animal Science of Chinese Academy of Medical Sciences (Beijing, China). For intracoronary delivery of adenoviruses, the mice were anesthetized and ventilated with a HX-300S animal ventilator (Chengdu, China). The entry to the chest was through a small left anterior thoracotomy, the pericardial sac was then removed and 2 × 1010 MOI adenoviruses of CypD-siRNA or 2 × 1010 MOI adenoviruses of CypD-sc were injected with a catheter from the apex of the left ventricle into the aortic root while the aorta and pulmonary arteries were cross-clamped. The clamp was maintained for 20 s when the heart pumped against a closed system. After removal of air and blood, the chest was then closed, and the mice were returned back to cage for recovery. Five days after the injection of adenoviruses, the mice were subjected to I/R surgery.
For I/R injury model, mice were subjected to 60 min ischemia, then 3-h or 1-week reperfusion as described.49 Sham-operated group experienced the same procedure except the snare was left untied. miR-30b transgenic mice, E2F1 knockout mice and WT mice were subjected to I/R. The treatment with Evans blue dye was as described.49 The areas of infarction (INF), AAR and non-ischemic left ventricle (LV) were assessed with the computer-assisted planimetry (NIH Image 1.57, Bethesda, MD, USA) by an observer blinded to the sample identity. The ratio of AAR/LV and INF/AAR were calculated as described.49 All experiments were performed according to the protocols approved by the Institute Animal Care Committee.
Immunohistochemistry
CD31/TUNEL double staining was performed on 7-μm frozen sections. TUNEL staining was used to visualize apoptotic cell, while CD31 was used to identify endothelial cells in the mouse heart. Briefly, TUNEL staining was performed using the In situ Cell Death Detection Kit, FITC (Roche Applied Science, Mannheim, Germany) according to the manufacturer’s instructions. Anti-CD31/PECAM1 (Millipore, MA) was used as a primary antibody, followed by an anti-rabbit secondary antibody (TRITC). And slides were coverslipped using a mounting medium with DAPI to identify cell nuclei. Images were captured and processed using a laser-scanning confocal microscope (Zeiss LSM510 META, Oberkochen, Germany). Double-positive cells for CD31 and TUNEL were counted, and assessment was blinded to the treatment assignment.
Echocardiographic assessment
Transthoracic echocardiographic analysis was performed on mice after the sham or I/R surgery as we described.44 Echocardiographic parameters such as systolic left ventricular internal diameters (LVIDs) and diastolic left ventricular internal diameters (LVIDd) were measured. Fractional shortening of left ventricular diameter was calculated as ((LVIDd−LVIDs)/LVIDd) × 100. After in vivo evaluation of the cardiac function, the mice were euthanized, and the hearts were harvested, weighted and used for histological examination.
Statistical analysis
Data are expressed as the mean±S.E.M. of at least three independent experiments. We used a one-way analysis of variance for multiple comparisons. A value of P<0.05 was considered significant.
Abbreviations
- miRNA:
-
microRNA
- H2O2:
-
hydrogen peroxide
- PI:
-
propidium iodide
- CypD:
-
Cyclophilin D
- mPTP:
-
mitochondrial permeability transition pore
- I/R:
-
ischemia/reperfusion
- qRT-PCR:
-
quantitative reverse transcription-PCR
- TP:
-
target protector
References
Konstantinidis K, Whelan RS, Kitsis RN . Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol 2012; 32: 1552–1562.
Whelan RS, Kaplinskiy V, Kitsis RN . Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 2010; 72: 19–44.
Sosna J, Voigt S, Mathieu S, Lange A, Thon L, Davarnia P et al. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci 2014; 71: 331–348.
Murakami Y, Matsumoto H, Roh M, Giani A, Kataoka K, Morizane Y et al. Programmed necrosis, not apoptosis, is a key mediator of cell loss and DAMP-mediated inflammation in dsRNA-induced retinal degeneration. Cell Death Differ 2014; 21: 270–277.
Moujalled DM, Cook WD, Okamoto T, Murphy J, Lawlor KE, Vince JE et al. TNF can activate RIPK3 and cause programmed necrosis in the absence of RIPK1. Cell Death Dis 2013; 4: e465.
Moquin DM, McQuade T, Chan FK . CYLD deubiquitinates RIP1 in the TNFalpha-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS One 2013; 8: e76841.
Hamahata K, Adachi S, Matsubara H, Okada M, Imai T, Watanabe K et al. Mitochondrial dysfunction is related to necrosis-like programmed cell death induced by A23187 in CEM cells. Eur J Pharmacol 2005; 516: 187–196.
Baines CP . Role of the mitochondrion in programmed necrosis. Front Physiol 2010; 1: 156.
Kingma JG Jr., Yellon DM . Inability of dimethylthiourea to limit tissue necrosis during acute myocardial infarction in rabbits. Free Radic Biol Med 1992; 12: 263–270.
Lissoni P, Pelizzoni F, Mauri O, Perego M, Pittalis S, Barni S . Enhanced secretion of tumour necrosis factor in patients with myocardial infarction. Eur J Med 1992; 1: 277–280.
Bryant D, Becker L, Richardson J, Shelton J, Franco F, Peshock R et al. Cardiac failure in transgenic mice with myocardial expression of tumor necrosis factor-alpha. Circulation 1998; 97: 1375–1381.
Guerra S, Leri A, Wang X, Finato N, Di Loreto C, Beltrami CA et al. Myocyte death in the failing human heart is gender dependent. Circ Res 1999; 85: 856–866.
Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H et al. Ca2+− and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest 2007; 117: 2431–2444.
Kostin S, Pool L, Elsasser A, Hein S, Drexler HC, Arnon E et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res 2003; 92: 715–724.
Woodfield K, Ruck A, Brdiczka D, Halestrap A . Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J 1998; 336: 287–290.
Crompton M . On the involvement of mitochondrial intermembrane junctional complexes in apoptosis. Curr Med Chem 2003; 10: 1473–1484.
Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004; 427: 461–465.
Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD . Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 2007; 9: 550–555.
Jobe SM, Wilson KM, Leo L, Raimondi A, Molkentin JD, Lentz SR et al. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008; 111: 1257–1265.
Eguchi Y, Shimizu S, Tsujimoto Y . Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res 1997; 57: 1835–1840.
Crompton M, Ellinger H, Costi A . Inhibition by cyclosporin A of a Ca2+-dependent pore in heart mitochondria activated by inorganic phosphate and oxidative stress. Biochem J 1988; 255: 357–360.
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005; 434: 652–658.
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005; 434: 658–662.
Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003; 425: 415–419.
Ohtani K, Dimmeler S . Control of cardiovascular differentiation by microRNAs. Basic Res Cardiol 2011; 106: 5–11.
Liu J, van Mil A, Vrijsen K, Zhao J, Gao L, Metz CH et al. MicroRNA-155 prevents necrotic cell death in human cardiomyocyte progenitor cells via targeting RIP1. J Cell Mol Med 2011; 15: 1474–1482.
Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S et al. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J Clin Invest 2012; 122: 1222.
Field SJ, Tsai F-Y, Kuo F, Zubiaga AM, Kaelin WG Jr, Livingston DM et al. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 1996; 85: 549–561.
Johnson DG, Schwarz JK, Cress WD, Nevins JR . Expression of transcription factor E2f1 induces quiescent cells to enter S-phase. Nature 1993; 365: 349–352.
Hsieh MC, Das D, Sambandam N, Zhang MQ, Nahlé Z . Regulation of the PDK4 isozyme by the Rb-E2F1 complex. J Biol Chem 2008; 283: 27410–27417.
Cloud JE, Rogers C, Reza TL, Ziebold U, Stone JR, Picard MH et al. Mutant mouse models reveal the relative roles of E2F1 and E2F3 in vivo. Mol Cell Biol 2002; 22: 2663–2672.
Gardner AM, Xu FH, Fady C, Jacoby FJ, Duffey DC, Tu Y et al. Apoptotic vs. nonapoptotic cytotoxicity induced by hydrogen peroxide. Free Radic Biol Med 1997; 22: 73–83.
Takeda M, Shirato I, Kobayashi M, Endou H . Hydrogen peroxide induces necrosis, apoptosis, oncosis and apoptotic oncosis of mouse terminal proximal straight tubule cells. Nephron 1999; 81: 234–238.
Troyano A, Sancho P, Fernandez C, de Blas E, Bernardi P, Aller P . The selection between apoptosis and necrosis is differentially regulated in hydrogen peroxide-treated and glutathione-depleted human promonocytic cells. Cell Death Differ 2003; 10: 889–898.
Wei C, Li L, Gupta S . NF-kappaB-mediated miR-30b regulation in cardiomyocytes cell death by targeting Bcl-2. Mol Cell Biochem 2014; 387 (1-2): 135–141.
Stein A, Knodler M, Makowski M, Kuhnel S, Nekolla S, Keithahn A et al. Local erythropoietin and endothelial progenitor cells improve regional cardiac function in acute myocardial infarction. BMC Cardiovasc Disord 2010; 10: 43.
Quintavalle C, Donnarumma E, Iaboni M, Roscigno G, Garofalo M, Romano G et al. Effect of miR-21 and miR-30b/c on TRAIL-induced apoptosis in glioma cells. Oncogene 2013; 32: 4001–4008.
Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P . miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet 2010; 6: e1000795.
McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S . Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 2004; 286: H1923–H1935.
Angelis E, Zhao P, Zhang R, Goldhaber JI, Maclellan WR . The role of E2F-1 and downstream target genes in mediating ischemia/reperfusion injury in vivo. J Mol Cell Cardiol 2011; 51: 919–926.
Stiewe T, Pützer BM . Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat Genet 2000; 26: 464–469.
Tan WQ, Wang K, Lv DY, Li PF . Foxo3a inhibits cardiomyocyte hypertrophy through transactivating catalase. J Biol Chem 2008; 283: 29730–29739.
Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014; 40: 91–104.
Lin Z, Murtaza I, Wang K, Jiao J, Gao J, Li PF . miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc Natl Acad Sci USA 2009; 106: 12103–12108.
Li PF, Dietz R, von Harsdorf R . p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. EMBO J 1999; 18: 6027–6036.
Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 2005; 33: e179.
Lau P, Verrier JD, Nielsen JA, Johnson KR, Notterpek L, Hudson LD . Identification of dynamically regulated microRNA and mRNA networks in developing oligodendrocytes. J Neurosci 2008; 28: 11720–11730.
Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC . Autophagosome precursor maturation requires homotypic fusion. Cell 2011; 146: 303–317.
Wang JX, Jiao JQ, Li Q, Long B, Wang K, Liu JP et al. miR-499 regulates mitochondrial dynamics by targeting calcineurin and dynamin-related protein-1. Nat Med 2011; 17: 71–78.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81270160) and the Beijing Natural Science Foundation (7142103).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Wang, K., An, T., Zhou, LY. et al. E2F1-regulated miR-30b suppresses Cyclophilin D and protects heart from ischemia/reperfusion injury and necrotic cell death. Cell Death Differ 22, 743–754 (2015). https://doi.org/10.1038/cdd.2014.165
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2014.165
This article is cited by
-
Repositioning linifanib as a potent anti-necroptosis agent for sepsis
Cell Death Discovery (2023)
-
Non-Coding RNA-Mediated Gene Regulation in Cardiovascular Disorders: Current Insights and Future Directions
Journal of Cardiovascular Translational Research (2023)
-
Cell death regulation by MAMs: from molecular mechanisms to therapeutic implications in cardiovascular diseases
Cell Death & Disease (2022)
-
Targeting the pathways of regulated necrosis: a potential strategy for alleviation of cardio-cerebrovascular injury
Cellular and Molecular Life Sciences (2021)
-
Reciprocal regulation of miR-1205 and E2F1 modulates progression of laryngeal squamous cell carcinoma
Cell Death & Disease (2019)
