
Abstract
FLIP is a potential anti-cancer therapeutic target that inhibits apoptosis by blocking caspase 8 activation by death receptors. We report a novel interaction between FLIP and the DNA repair protein Ku70 that regulates FLIP protein stability by inhibiting its polyubiquitination. Furthermore, we found that the histone deacetylase (HDAC) inhibitor Vorinostat (SAHA) enhances the acetylation of Ku70, thereby disrupting the FLIP/Ku70 complex and triggering FLIP polyubiquitination and degradation by the proteasome. Using in vitro and in vivo colorectal cancer models, we further demonstrated that SAHA-induced apoptosis is dependant on FLIP downregulation and caspase 8 activation. In addition, an HDAC6-specific inhibitor Tubacin recapitulated the effects of SAHA, suggesting that HDAC6 is a key regulator of Ku70 acetylation and FLIP protein stability. Thus, HDAC inhibitors with anti-HDAC6 activity act as efficient post-transcriptional suppressors of FLIP expression and may, therefore, effectively act as ‘FLIP inhibitors’.
Similar content being viewed by others


Main
FLIP is an anti-apoptotic protein that blocks the activation of apoptosis mediated by death receptors, such as Fas, TRAIL receptor 1 (TRAIL-R1/DR4) and TRAIL-R2 (DR5).1 By binding to the adaptor protein FADD, FLIP inhibits apoptosis by blocking the processing and activation of procaspase 8 (FLICE) by death receptor complexes termed DISCs (death-inducing signalling complexes).2 We previously reported that FLIP inhibits apoptosis induced by chemotherapeutic agents3 and that high FLIP expression is an independent adverse prognostic biomarker in colorectal cancer (CRC).4 These and other studies have indicated that inhibition of FLIP constitutes a promising therapeutic strategy for the treatment of CRC.
Ku70 and its binding partner Ku80 are critical components of the non-homologous end joining (NHEJ) DNA repair machinery.5 Ku70 is regulated by acetylation, which is mediated by the histone acetyl transferases (HATs); CREB-binding protein (CBP) and PCAF, and its acetylation can be enhanced by treating cells with histone deacetylase (HDAC) inhibitors.6 Ku70 acetylation disrupts its DNA-binding activity and sensitises cells to DNA-damaging agents.7 In addition, cytoplasmic Ku70 binds to and regulates the pro-apoptotic Bcl-2 family member, Bax.6 Ku70 simultaneously inhibits Bax degradation via the ubiquitin proteasome system (UPS) and prevents its translocation to the mitochondria.8 Moreover, it has been reported that Ku70 may have intrinsic de-ubiquitinating (DUB) activity.8 The Ku70–Bax complex is disrupted by Ku70 acetylation, which promotes Bax translocation to mitochondria and apoptosis induction.6
Herein, we report a novel interaction between FLIP and Ku70 that regulates FLIP stability. This interaction is acetylation-dependant and is disrupted by HDAC inhibitors with activity against HDAC6. Disruption of the Ku70–FLIP interaction subsequently leads to FLIP degradation by the UPS and induction of caspase 8-dependant apoptosis.
Results
FLIP interacts with Ku70
A yeast-2-hybrid screen was carried out to identify novel binding partners for the long FLIP splice variant (FLIP(L)). One of the novel proteins identified was Ku70. The interaction between FLIP(L) and Ku70 was confirmed by re-transformation assay (Figure 1a). Immunoprecipitation experiments confirmed that FLIP(L) interacts with Ku70 in CRC cells (Figure 1b). Despite the predominant site of Ku70 expression being nuclear, the interaction between FLIP(L) and Ku70 was more prominent in cytosolic fractions (Figure 1c), suggesting that the primary site of the interaction is cytosolic. Ku70 also interacted with the short FLIP splice form, FLIP(S) (Supplementary Figure S1A). To map Ku70's interaction with FLIP, we generated a series of Flag-tagged Ku70 constructs. The region from 430 to 609 amino acids interacted strongly with FLIP, however, neither the 496–609 nor the 535–609 fragments interacted (Figure 1d). These mapping experiments indicate that Ku70 binds FLIP via a region between amino acids 430 and 496, which encompasses one of its sites of interaction with Ku80.

FLIP interacts with Ku70. (a) Yeast-2-hybrid re-transformation assay: blue colonies are visible when pGADT7Rec-Ku70 was co-transfected with pGBKT7-FLIP(L), but not pGBKT7-empty vector (EV) indicating that Ku70 and FLIP(L) interact. (b) Lysates from HCT116 cells transfected with Flag-tagged FLIP(L) were immunoprecipitated (IP) with anti-Ku70 or control IgG antibodies and then immunoblotted (IB) for the presence of Flag-tagged FLIP(L). (c) Nuclear (Nuc) and cytosolic (Cyt) fractions were prepared from HCT116 cells, and equal amounts of protein were immunoprecipitated (IP) with anti-FLIP (H202) antibody and then immunoblotted (IB) for Ku70 and FLIP. Short (2.5′) and long (10′) exposures are presented and the relative levels of FLIP and Ku70 in the inputs are also shown. (d) Schematic diagram of Ku70 and Flag-tagged Ku70 expression constructs. Protein lysates from HCT116 cells transfected with Flag-tagged Ku70 constructs were immunoprecipitated with anti-FLIP (H202) antibody and then immunoblotted for Flag-tagged Ku70. Successful immunoprecipitation of FLIP was confirmed using the anti-FLIP antibody, NF6. The color reproduction of this figure is available at the Cell Death and Differentiation journal online
Ku70 binds to DED2 of FLIP
To identify the regions of FLIP that are important for the FLIP–Ku70 interaction, we used a FLIP peptide array of overlapping 25-mer peptides, each shifted by four amino acids, encompassing the entire FLIP(S) sequence.9 Duplicate membranes were overlaid with lysates from cells transfected with Flag-Ku70, and the Flag epitope detected by immunoblotting. Strong binding of Ku70 to the array was detected in four regions (Figure 2a and table). To map the FLIP–Ku70 interaction to specific amino acids, alanine substitution arrays were constructed for peptides 17, 30, 41 and 46. Several alanine substitutions in peptide 30 (but not the other 3 peptides) were found to disrupt the binding of Ku70 (Figure 2b). By using viral FLIP MC159 as a template,10 a homology model of human FLIP(S) was generated. The model predicted that two of the candidate residues identified in the alanine substitution array, Y119 and R122, are prominently surface exposed and, therefore, available for making protein–protein interactions (Figure 2c); the other candidate residues were predicted to be buried. Mutation of FLIP(S) R122, but not Y119, to alanine significantly diminished binding to Ku70 (Figure 2d). Thus, these studies identify R122 as a critical site of interaction between FLIP and Ku70. This region of FLIP death effector domain 2 (DED2) is present in all FLIP splice forms and is adjacent to the ‘hydrophobic patch’ region reported to be important for binding to FADD.11 Binding of FADD and procaspase 8 to FLIP was unaffected by R122A mutation indicating that FADD/procaspase 8 and Ku70 interact with distinct regions of FLIP (Figure 2d).

Mapping the Ku70-binding site on FLIP. (a) Array of overlapping 25-mer peptides each shifted by four amino acids along the FLIP(S) protein sequence. The array was overlaid with lysates from EV or Flag-Ku70 transfected HCT116 cells. Positively interacting peptides generate dark spots. Four potential Ku70-binding regions were identified and are listed in the table. (b) An alanine scan of peptide 30, in which each residue was sequentially replaced by alanine, was overlaid with lysate from Flag-Ku70-transfected HCT116 cells. Loss of signal compared with wild-type (WT) control was observed for several residues. (c) Homology model of FLIP(S) highlighting the surface exposed R122 and Y119 residues. (d) GST pull-down demonstrating that FLIP R122A mutation, but not Y119A mutation, results in the loss of interaction with Ku70; neither mutation affects the interaction with FADD or procaspase 8
SAHA disrupts the FLIP–Ku70 interaction and induces FLIP degradation
Treatment with the HDAC inhibitor Vorinostat (SAHA, suberoylanilide hydroxamic acid) for 2 h disrupted the interaction between endogenous FLIP and Ku70 (Figure 3a), and increased Ku70 acetylation after 1 h (Figure 3b). Increased Ku70 acetylation following SAHA treatment was also observed in three other CRC cell lines (Supplementary Figure S1B). Although Ku70 was primarily expressed in the nucleus, acetylated Ku70 was detectable at similar levels in the nucleus and the cytosol, and increased in both sub-cellular locations following SAHA treatment (Supplementary Figure S1C). We were unable to detect the acetylation of FLIP following SAHA treatment (Supplementary Figure S1D), suggesting that it is not directly modified by acetylation. Ku70 acetylation following SAHA treatment decreased its capacity to bind to FLIP (Figure 3b). Cohen et al.6 demonstrated that mimicking the acetylation of lysine residues K539 or K542 in Ku70 by mutating them to glutamine disrupted its ability to inhibit Bax-mediated apoptosis. We found that mutation of either of these residues to glutamine impaired the ability of Ku70 to interact with FLIP (Figure 3c). Collectively, these results indicate that Ku70 acetylation inhibits its ability to interact with FLIP.

SAHA disrupts the FLIP–Ku70 complex and induces FLIP degradation. (a) Protein lysates from HCT116 cells treated with 1 μM SAHA were immunoprecipitated (IP) with anti-FLIP (H202) or control IgG antibodies, and then immunoblotted (IB) for the presence of Ku70. Immunoprecipitation of FLIP was confirmed using the anti-FLIP antibody, NF6. (b) Protein lysates from HCT116 cells treated with 1 μM SAHA for 1–2 h were either immunoprecipitated (IP) with anti-acetylated lysine (AcK) antibody (top panel) or incubated with GST-FLIP(S) protein (middle panel). The resulting protein complexes were then immunoblotted (IB) for FLIP and Ku70. 5% of the indicated lysates were immunoblotted for the presence of Ku70. (c) Flag-tagged wild-type (WT), K539Q and K542Q mutant expression constructs were transfected into HCT116 cells, and the protein lysates were immunoprecipitated (IP) with anti-FLIP (H202) and immunoblotted with anti-Flag antibody. Immunoprecipitation of FLIP was confirmed using the anti-FLIP antibody, NF6. (d) Flag-FLIP(L)/FLIP(S) vectors (2 μg) were transfected into HCT116 cells for 24 h. Cells were then treated with 1 μM SAHA or 10 μM MG132 for 4 h. Cell lysates were immunoprecipitated with anti-polyubiquitin antibody and immunoblotted for Flag-tagged FLIP. (e) Western blot analysis of HCT116 cells treated with 1 μM SAHA. FLIP and Ku70 expression and PARP cleavage (a hallmark of apoptosis) were analysed by immunoblotting
Disruption of FLIP–Ku70 binding following SAHA treatment correlated with increased polyubiquitination of FLIP(L) and FLIP(S) (Figure 3d) and rapid downregulation of both splice forms from ∼4 h following treatment (Figure 3e). SAHA treatment also downregulated FLIP protein expression in three other CRC cell lines after 6 h at sub-IC50 doses (Supplementary Figures S2C and E). In contrast to other studies, which have found that HDAC inhibitors downregulate FLIP at the transcriptional level,12, 13 we found no evidence of transcriptional suppression of FLIP in CRC cells (Supplementary Figures S2A and D). Moreover, co-treatment with the proteasome inhibitor MG132 attenuated SAHA-induced FLIP downregulation (Supplementary Figure S2B), providing further evidence that FLIP downregulation is mediated by the UPS.
Ku70 post-transcriptionally regulates FLIP expression
Transfection with three different Ku70-targeted siRNAs resulted in Ku70 depletion and significant decreases in FLIP expression (Figure 4a). Similar effects were observed in three other CRC cell lines (Figure 4b). However, Ku70 depletion had no effect on FLIP mRNA expression (Figure 4c). siRNA-mediated Ku70 downregulation resulted in apoptosis as indicated by poly(ADP-ribose) polymerase (PARP) cleavage (Figure 4d), flow cytometry (Figure 4e) and caspase 3/7 activation (Supplementary Figure S3A). The FLIP downregulation observed in Ku70-depleted cells was not an indirect result of caspase activation, as inhibition of apoptosis with the pan-caspase inhibitor z-VAD-fmk failed to prevent FLIP downregulation in Ku70-depleted cells (Figure 4d). Furthermore, the half-lives of both FLIP(L) and FLIP(S) were significantly reduced in Ku70-depleted cells, with almost undetectable levels of FLIP 1 h after treatment with the protein synthesis inhibitor cycloheximide (Figure 4f). Importantly, Ku70 downregulation resulted in increased levels of polyubiquitinated FLIP(L) and FLIP(S) (Figure 4g), suggesting that Ku70 enhances the half-life of FLIP by inhibiting its degradation via the UPS.

Ku70 regulates FLIP protein stability. (a) HCT116 cells were transfected for 48 h with 50 nM control (SC) or three different Ku70-targeted (siKu#2–4) siRNAs, and FLIP and Ku70 expression were analysed by immunoblotting. (b) HCT116, HT29, H630 and RKO CRC cells were transfected for 48 h with 50 nM SC or siKu, and FLIP and Ku70 expression were analysed by immunoblotting. Densitometry was used to calculate the ratio of the expression of each FLIP splice form in siKu-transfected cells compared with SC-transfected cells after normalisation to Actin. (c) TaqMan Q-PCR analysis of FLIP mRNA expression in HCT116 cells transfected with 50 nM SC or siKu#2 for 48 h. Relative mRNA expression was calculated using the ΔΔCt method with normalisation to GAPDH. (d) Western blot analysis of FLIP and Ku70 expression in HCT116 cells transfected with 50 nM SC or siKu#2 for 48 h; cells were co-treated with 10 μM pan-caspase inhibitor, z-VAD-fmk. (e) HCT116 parental and Bax-null cells were transfected with 50 nM SC or Ku70 siRNAs (siKu) for 48 h. Cells were co-transfected with 20 nM siCasp8 as indicated, and apoptosis was determined by flow cytometry (*P<0.05; Student's t-test). (f) HCT116 cells transfected with 50 nM SC or siKu#2 for 48 h were treated with 100 μg/ml cyclohexamide (CHX) before analysis of FLIP expression by western blot. Densitometry was used to estimate the half-life (t1/2) of each FLIP splice form after normalisation of expression to Actin. (g) Flag-FLIP(L)/FLIP(S) vectors (2 μg) were co-transfected into HCT116 cells with 50 nM SC or siKu#2 for 48 h. Cell lysates were immunoprecipitated with anti-polyubiquitin antibody and immunoblotted for Flag-tagged FLIP
SAHA- and Ku70 siRNA-induced apoptosis is caspase 8-dependant
Analysis of other apoptotic proteins indicated that unlike FLIP, none were significantly affected by SAHA treatment after 6 h, however, Bax was upregulated and XIAP downregulated 24 h following SAHA treatment (Figure 5a). Despite being acetylated, total Ku70 expression was not affected by SAHA treatment (Figure 5a). Expression of several BH3-only proteins was also assessed: NOXA and BID were not significantly altered following SAHA treatment, whereas PUMA was downregulated and BIM upregulated (Figure 5b). As the upregulation of Bax and BIM at 24 h coincided with the onset of apoptosis as determined by PARP cleavage, we directly assessed the roles of the initiator caspases, caspase 8 and caspase 9 during SAHA-induced apoptosis. siRNA-mediated inhibition of caspase 8 significantly inhibited SAHA-induced apoptosis in each CRC cell line as assessed by flow cytometry (Figure 5c), PARP cleavage (Supplementary Figure S4A) and analysis of caspase 3/7 activity (Supplementary Figure S4C). In contrast, caspase 9 silencing failed to inhibit SAHA-induced apoptosis (Figure 5c). Furthermore, caspase 8 activity increased following SAHA treatment and correlated with increased activity of the executioner caspases 3/7 (Figure 5d). SAHA treatment also resulted in significant increases in caspase 8 activity in other CRC cell lines (Supplementary Figure S4B). Notably, increased caspase 8 activity was detected in Ku70-depleted HCT116 cells (Supplementary Figure S3A), and the apoptosis induced by Ku70 silencing was caspase 8-dependant (Figure 4e). Similar results were observed in HT29 and RKO cells, although not in H630 cells, in which Ku70 silencing failed to activate either caspase 8 or caspase 3/7 (Supplementary Figures S3B and S3C); this may be because FLIP downregulation following Ku70 depletion was less pronounced in this cell line (Figure 4b). Interestingly, given Ku70's role in regulating Bax-mediated apoptosis,6 Ku70 depletion still resulted in significant (albeit reduced) levels of apoptosis in Bax-null HCT116 cells, and this was again caspase 8-dependant (Figure 4e).

SAHA-induced apoptosis is caspase-8 dependant. (a) HCT116 cells were treated with SAHA for 6 and 24 h, and FLIP, procaspase 8, XIAP, Bcl-2, Mcl-1, Bcl-XL, Bax, Bak and Ku70 expression and PARP cleavage were analysed by immunoblotting. (b) HCT116 cells were treated with SAHA for 24 h, and FLIP, PUMA, NOXA, BIM and BID expression were analysed by immunoblotting. (c) HCT116 cells were transfected with 20 nM SC, caspase 8-targeted (siCasp8) or caspase 9 (siCasp9)-targeted siRNAs for 24 h followed by treatment with 1 μM SAHA for a further 24 h. Apoptosis was determined by assessing the percentage of cells in the sub-G0/G1 apoptotic fraction. (d) HCT116 cells were treated with increasing doses of SAHA for 24 h and caspase 3/7-like (DEVD-ase) and 8-like (IETD-ase) activity were measured using Caspase-Glo assays (*P<0.05; **P<0.01; ***P<0.001; Student's t-test)
SAHA- and Ku70 siRNA-induced apoptosis is FLIP-dependant
Using an HCT116 FLIP(L) overexpressing cell line (FL17), we obtained direct evidence for the importance of FLIP downregulation during SAHA-induced apoptosis. SAHA failed to downregulate FLIP(L) in this cell line (Figure 6a) as it activated transcription of the exogenous transgene (unpublished observations). Lack of FLIP(L) downregulation in this cell line correlated with significantly reduced levels of SAHA-induced apoptosis compared with the parental cell line as determined by PARP cleavage (Figure 6a) and flow cytometry (Figure 6b). Of note, FLIP(L) overexpression did not prevent SAHA-induced cell cycle arrest in G2/M phase (Figure 6b), indicating that FLIP regulates the cytotoxic rather than the cytostatic effects of SAHA. SAHA also sensitised CRC cells to apoptosis induced by rTRAIL and chemotherapeutic agents in a FLIP-dependant manner (Supplementary Figures S5A-D). These results establish direct evidence for FLIP downregulation as a major mechanism of apoptosis induction by SAHA alone and in combination with other anti-cancer agents. Ku70 siRNA-induced apoptosis was also significantly reduced in FLIP(L)-overexpressing cells (Figure 6c).

Apoptosis induced by Vorinostat is regulated by FLIP both in vitro and in vivo. (a) HCT116 parental and FLIP(L) overexpressing (FL17) cells were treated with SAHA for 24 h and analysed by western blot for PARP cleavage and expression of FLIP and Procaspase-8. (b) Cell cycle analysis of parental and FL17 cells treated with 2.5 μM SAHA for 24 and 48 h; the sub-G1 apoptotic fraction in SAHA-treated cells is given. (c) Flow cytometric analysis of apoptosis in parental and FL17 cells transfected with 50 nM control (SC) or Ku70 siRNA (siKu70) for 48 h. (d) Western blot analysis of FLIP(L) and acetylated histone H4 expression in parental and FL17 HCT116 xenografts from mice injected IP with 100 mg/kg SAHA or vehicle for 2 weeks using a 5-day on/2-day off schedule. Xenografts were harvested 6 h after the final treatment. (e) Growth of HCT116 and FL17 xenografts in mice injected IP with 100 mg/kg SAHA (**P<0.01; ***P<0.001; Student's t-test)
To assess the importance of FLIP downregulation for the in vivo effects of SAHA, we used the FL17 FLIP(L)-overexpressing model. In both parental and FL17 xenografts, treatment with SAHA resulted in increased acetylation of histone H4, a marker of HDAC inhibition (Figure 6d). SAHA treatment resulted in potent FLIP(L) downregulation in the parental xenografts and also decreased FLIP(L) expression in the FL17 xenografts. However, the levels of FLIP(L) expression that were maintained in SAHA-treated FL17 tumours were comparable to those in vehicle-treated parental xenografts. Notably, SAHA treatment failed to inhibit the growth of FL17 xenografts, whereas the growth of the parental xenografts was significantly inhibited by SAHA (Figure 6e). These results are the first to demonstrate that SAHA downregulates FLIP in vivo and that FLIP downregulation is important for its anti-tumour activity.
Inhibitors with anti-HDAC6 activity regulate FLIP expression and Ku70 acetylation
To identify the HDACs involved in regulating Ku70 acetylation, we used the HDAC inhibitor Droxinostat,14 which, although less potent, has a more restricted activity than SAHA: Droxinostat is an inhibitor of HDACs 6 and 8, and (to a lesser extent) HDAC3;14 SAHA inhibits HDACs 1, 2, 3, 6 and (to a lesser extent) 8.15 In agreement with earlier studies,16 we found that Droxinostat potently downregulated FLIP expression and did so at early (6 h) time points post-treatment (Supplementary Figure S6A). However, unlike these earlier studies, we found that this downregulation was post-transcriptional, as this agent had no effect on FLIP mRNA expression (Supplementary Figure S6B). Droxinostat-induced apoptosis correlated with FLIP downregulation (Supplementary Figure S6C), and FLIP downregulation was not caspase dependant (Supplementary Figure S6D). Like SAHA, Droxinostat-induced apoptosis was caspase 8-dependant (Supplementary Figures S6D and 7A). Using a ‘caspase-trap’ assay,17 we demonstrated that caspase 8 rather than caspase 9 was the first caspase activated in HCT116 cells in response to treatment with both SAHA and Droxinostat for 4 h (Figure 7b). In addition, Droxinostat-induced apoptosis was inhibited in FL17 cells (Figures 7c and d). Given the reported importance of Bax for HDAC inhibitor-induced apoptosis,6 it was notable that the inhibitory effects of caspase 8 silencing or FLIP overexpression on HDAC inhibitor-induced apoptosis were more profound than loss of Bax (Figures 7a and d). Furthermore, in contrast to a previous study,18 we found that HDAC inhibitor-induced apoptosis was not p53-dependant in HCT116 cells (Figure 7d). We previously reported that apoptosis induction following siRNA-mediated FLIP silencing is primarily driven by the TRAIL receptor DR5.3 In agreement with this, SAHA- and Droxinostat-induced cell death were both significantly attenuated when DR5 was silenced, and this effect was further enhanced when DR4 was co-silenced with DR5 (Figure 7e).

The HDAC3, 6 and 8 inhibitor downregulates FLIP expression. (a) HCT116 parental and Bax-null cells were transfected with 10 nM SC or caspase 8-targeted (siCasp8) siRNAs for 24 h followed by treatment with 1 μM SAHA or 30 μM Droxinostat (Drox) for a further 24 h. Apoptosis was determined by flow cytometry by assessing the percentage of propidium iodide-stained cells in the sub-G0/G1 apoptotic fraction. (b) HCT116 cells were pre-incubated in the presence of 50 μM biotinylated VAD–fmk for 2 h and then treated with either 10 ng/ml TRAIL, 30 μM Droxinostat or 1 μM SAHA and cultured for a further 4 h. Cells were then lysed and active caspases precipitated using streptavidin–agarose and identified by immunoblotting using specific caspase antibodies. (c) HCT116 parental and FLIP(L) overexpressing (FL17) cells were treated with 10, 30 and 60 μM Droxinostat (Drox) for 24 h, and the resulting cell lysates were separated by SDS-PAGE and immunoblotted for FLIP(L) and FLIP(S). (d) HCT116 parental, Bax-null, p53-null and FL17 cells were treated with 1 μM SAHA or 30 μM Drox for 24 h, and apoptosis was determined by flow cytometry by assessing the percentage of propidium iodide-stained cells in the sub-G0/G1 apoptotic fraction. (e) HCT116 parental cells were transfected with 10 nM control (SC), DR4- or DR5-targeted siRNAs for 24 h followed by treatment with 1 μM SAHA or 30 μM Drox for a further 24 h. Apoptosis was determined by flow cytometry by assessing the percentage of propidium iodide-stained cells in the sub-G0/G1 apoptotic fraction (ns, not significant; *P<0.05; **P<0.01; ***P<0.001; Student's t-test)
As Droxinostat and SAHA both inhibit HDAC6, we hypothesised that HDAC6 may be involved in regulating FLIP expression. To test this, we used an HDAC6-selective inhibitor Tubacin.19 Both Tubacin and Droxinostat increased Ku70 acetylation to the same extent as SAHA (Figure 8a). By comparison, acetylation of Ku70 in response to Entinostat, which inhibits HDACs 1–3, but not HDAC6,15 was significantly less than that induced by the other HDAC inhibitors (Figure 8a). In addition, siRNA-mediated depletion of HDAC6 resulted in significantly increased Ku70 acetylation (Figure 8b). Like SAHA and Droxinostat, Tubacin treatment resulted in rapid FLIP protein downregulation (Figure 8c), but had no significant effect on FLIP mRNA expression (Figure 8d). Tubacin also caused an increase in acetylation of α-tubulin, a marker of HDAC6 inhibition, whereas acetylation of histone H4, a marker of nuclear HDAC inhibition, was significantly lower following Tubacin treatment than following SAHA treatment (Figure 8c). In contrast, Entinostat failed to downregulate FLIP expression in HCT116 cells after 6 h at a concentration that potently increased acetylation of histone H4, but had no effect on acetylation of α-tubulin (Figure 8e). In addition, Tubacin-induced apoptosis was highly FLIP-dependant as determined by flow cytometry (Figure 8f), consistent with FLIP downregulation being a primary mechanism of apoptosis induction in response to this agent.

The HDAC6 inhibitor Tubacin acetylates Ku70 and downregulates FLIP. (a) Immunoprecipitation reactions examining the effect of 2 h treatment of HCT116 cells with 1 μM SAHA, 30 μM Droxinostat (DROX), 5 μM Tubacin (TUBA) and 1 μM Entinostat (ENT) on acetylation of Ku70. Lysates were immunoprecipitated with anti-acetylated lysine (AcK) antibody and immunoblotted for Ku70. (b) Immunoprecipitation reactions examining the effect of siRNA-mediated depletion of HDAC6 on the acetylation of Ku70. HCT116 cells were transfected with 5 nM HDAC6-targeted siRNA (siHDAC6) for 48 h. Lysates were prepared, immunoprecipitated with anti-acetylated lysine (AcK) antibody and immunoblotted for Ku70. Inputs confirmed equal levels of total Ku70 and downregulation of HDAC6 following transfection with siHDAC6. (c) Western blot analysis of expression of FLIP, acetylated histone H4 and acetylated α-tubulin in HCT116 cells 6 h after treatment with 1 μM SAHA, or 2.5 or 5 μM Tubacin. (d) Q-PCR analysis of FLIP mRNA expression in HCT116 cells treated with 5 μM Tubacin for 6 h. (e) Western blot analysis of expression of FLIP, acetylated histone H4 and acetylated α-tubulin in HCT116 cells 6 h after treatment with 1 μM Entinostat. (f) HCT116 parental and FLIP(L) overexpressing (FL17) cells were treated with 5 μM Tubacin for 24 h, and apoptosis was measured by flow cytometry analysis of Annexin V stained cells (*P<0.05; Student's t-test)
Discussion
We previously demonstrated that FLIP is an important regulator of apoptosis induction and drug resistance in CRC. 3, 20 We carried out a yeast-2-hybrid screen to identify novel FLIP-interacting proteins, with the aim of identifying novel regulators of FLIP expression or function. This screen identified Ku70, a DNA repair protein that was initially characterised as being critical for NHEJ.21 Ku70 was subsequently shown to have an apoptosis-regulating function through its ability to bind to and sequester Bax.6 Both the DNA- and Bax-binding activities of Ku70 are acetylation dependant.6, 7 Acetylation of Ku70 at residues K539 and K542 in its C-terminus by CBP and PCAF disrupt its ability to inhibit Bax-mediated apoptosis.6 Moreover, Ku70 was reported to have intrinsic DUB activity that inhibits Bax degradation via the UPS.8
We found that FLIP interacts with Ku70 via a region in the DED2, using a site (R122) adjacent to the region that is functionally important for FLIP binding to FADD.11 Binding of FADD and procaspase 8 to FLIP was unaffected by mutation of R122, indicating that Ku70 interacts with a region of FLIP that is distinct from the region with which it interacts with these DISC components. Furthermore, Ku70 did not bind to FLIP via the same region it binds Bax, but rather via a region previously shown to be important for dimerisation with Ku80 (amino acids 430–496).22 Therefore, FLIP interacts with the same region of Ku70 with which it engages Ku80 and does so using a region in DED2 not involved in its DISC recruitment. In addition, fractionation experiments suggested that similar to Bax, the primary sub-cellular location of the FLIP–Ku70 interaction is cytoplasmic.
Ku70 depletion resulted in significant decreases in FLIP expression. Furthermore, the decrease in FLIP expression following Ku70 silencing was because of a decrease in protein stability. Treatment with the HDAC inhibitor SAHA resulted in the acetylation of Ku70 and disrupted its interaction with FLIP. Moreover, mimicking Ku70 acetylation by mutating the residues K539 and K542 to glutamine resulted in reduced Ku70–FLIP binding. These lysine residues are present in the flexible C-terminal linker region adjacent to the region of Ku70, which binds FLIP (amino acids 430–496). It is possible that acetylation of this flexible linker region causes a conformational change of Ku70 that disrupts binding of FLIP in the adjacent 430–496 region, or that acetylation disrupts binding of an as yet unidentified binding partner (or partners) that is important for maintaining the Ku70–FLIP complex.
Although we found no evidence of FLIP acetylation following SAHA treatment, polyubiquitination of both FLIP(L) and FLIP(S) was increased, and both splice forms were rapidly downregulated in a proteasome-dependant manner. A direct link between Ku70 and FLIP ubiquitination status was indicated by the increased levels of polyubiquitinated FLIP(L) and FLIP(S) in Ku70-silenced cells. In contrast to numerous other studies,12, 13 we found no evidence that SAHA downregulated FLIP expression at the transcriptional level. However, our findings agree with a recent study in breast cancer models, which also found that SAHA-induced FLIP downregulation was post-transcriptional.23 In addition, a related pan-HDAC inhibitor Panobinostat (LBH589) was recently reported to cause FLIP polyubiquitination and proteasomal degradation in pancreatic cancer cells.24 Collectively, these findings suggest that SAHA causes Ku70 acetylation, resulting in the disruption of its interaction with FLIP leading to degradation of FLIP by the UPS.
Although Ku70 is a substrate for the HATs CBP and PCAF,6 the HDACs that regulate Ku70 acetylation have not been previously characterised. In this study, we provide evidence that HDAC6 is a key deacetylase for Ku70 as, in addition to Vorinostat (inhibitor of HDAC1-3, 6 and 8)15 and Droxinostat (inhibitor of HDAC3, 6 and 8),14 the HDAC6-specific inhibitor Tubacin19 potently increased the acetylation of Ku70. Moreover, both Droxinostat and Tubacin also caused rapid FLIP downregulation. In contrast, Entinostat, which inhibits HDAC1–3, but has no activity against HDAC6,15 failed to downregulate FLIP at early time points and had less impact on Ku70 acetylation. During revision of this manuscript, another study was published that also found that HDAC6 deacetylates Ku70.25 HDAC6 is a unique HDAC that has key roles in regulating protein turnover by the proteasome through its regulation of HSP90 and by the aggresome through its ability to recruit polyubiquitinated proteins to the tubulin cytoskeleton.26 Thus, this study identifies a further potential role for HDAC6 in regulating protein stability through its ability to deacetylate Ku70, thereby stabilising FLIP. By deacetylating Ku70, HDAC6 may also regulate Bax and possibly other as yet unidentified Ku70-binding partners.
Although a number of studies have shown that HDAC inhibitor-mediated downregulation of FLIP sensitises various cancer cell lines to death ligands, such as TRAIL,23, 27, 28 to our knowledge, no previous studies have directly examined whether FLIP downregulation is a sufficient death signal for apoptosis induction when these agents are administered as single agents. We found that FLIP downregulation in response to HDAC inhibitors results in caspase 8- and DR5-mediated apoptosis, phenocopying the mechanism of apoptosis that we previously observed in CRC cells following siRNA-mediated downregulation of FLIP.3 We also demonstrate for the first time that SAHA downregulates FLIP in vivo and that FLIP(L) overexpression abrogates SAHA's in vivo anti-tumour activity. FLIP(L) overexpression or caspase 8 downregulation had a greater inhibitory effect on HDAC inhibitor-induced apoptosis than loss of Bax, suggesting that the extrinsic apoptotic pathway is at least as important as the intrinsic pathway in mediating the effects of HDAC inhibitors in CRC cells.
From a cancer therapeutics perspective, these results suggest that HDAC inhibitors with anti-HDAC6 activity act as efficient post-transcriptional suppressors of FLIP expression in CRC. HDAC inhibitors are promising anti-cancer therapeutics that have demonstrated pre-clinical and clinical activities in haematological and solid cancers, and SAHA was the first of this class of compound to be FDA approved.29, 30, 31, 32, 33 There have been several early phase clinical trials in advanced, chemotherapy-resistant CRC examining SAHA in combination with standard 5-Fluorouracil (5-FU)-based chemotherapy.34, 35, 36 In one of these studies, 21/38 5-FU-refractory patients had stable disease and 1 had a partial response.34 The plasma concentrations of SAHA achieved in these patients were similar to the concentrations that we find are sufficient to downregulate FLIP. Of note, we found that SAHA synergized with both 5-FU and oxaliplatin to induce apoptosis in CRC cells; however, this synergy was abolished in cells overexpressing FLIP(L). Thus, FLIP and components of the extrinsic pathway, such as caspase 8, may be useful predictive biomarkers for the targeted use of HDAC inhibitors in CRC.
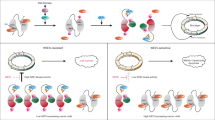
In conclusion, we provide evidence that Ku70 regulates FLIP ubiquitination and that HDAC inhibitor-mediated acetylation of Ku70 disrupts the interaction between FLIP and Ku70 resulting in degradation of FLIP via the UPS and induction of caspase 8-dependant apoptosis (summarised in Figure 9).

Schematic overview of main findings. Ku70 binds to and stabilizes FLIP by inhibiting its polyubiquitination. This maintains FLIP expression at a level at which it can prevent caspase 8 activation by complexes containing the adaptor protein FADD such as those formed after death receptor activation. HDAC inhibitors, particularly those that target HDAC6 such as SAHA, Droxinostat and Tubacin cause increased acetylation of Ku70, and this disrupts the FLIP/Ku70 complex, resulting in FLIP polyubiquitination and degradation via the proteasome. In the absence of FLIP, caspase 8 homodimers can be brought together by FADD, resulting in caspase 8 activation and apoptosis induction
Materials and Methods
Reagents
The following reagents were used: Vorinostat (Zolinza, SAHA; Selleck Chemicals, Suffolk, UK); Droxinostat (4-(4-chloro-2-methylphenoxy)-N-hydroxybutanamide, compound #5809354) and its inactive analogue 4-(4-chloro-2-methylphenoxy)-N-(3-ethoxypropyl)butanamide (compound #7271570, ChemBridge Corporation, San Diego, CA, USA). Tubacin was a kind gift from Dr. Ralph Mazitschek and Dr Stuart Schreiber (Howard Hughes Medical Institute, Broad Institute of Harvard and MIT). Entinostat (MS-275) was synthesised in the laboratory as previously described.37, 38 Cycloheximide and MG132 (Sigma-Aldrich, Dorset, UK). Biotinylated z-VAD-fmk (Val-Ala-Asp-CH2F) and immobilised Streptavidin slurry were purchased from MP Biomedicals (Cambridge, UK) and Thermo Fisher Scientific (Leicestershire, UK), respectively.
Cell culture
HCT116, H630, HT29 and RKO human CRC cell lines were used as previously described.3, 39 Matched isogenic p53 and Bax-null HCT116 cell lines were kindly provided by Professor B Vogelstein (Johns Hopkins University School of Medicine, Baltimore). FL17 cells were generated and cultured as previously described.20
Expression constructs
Flag-tagged FLIP(L), FLIP(S) and Ku70 expression constructswere generated in the pCMV-3Tag-6 vector (Agilent Technologies, Berkshire, UK). The GST-tagged FLIP(S) expression construct was generated in the pGEX6P-3 vector (GE Healthcare, Herts, UK). Mutagenesis was carried out using the KOD Extreme polymerase (Novagen, Nottingham, UK), with the template plasmid digested using DpnI (New England Biolabs, Herts, UK).
Western blotting and sub-cellular fractionation
Western blots were carried out as previously described.3 Isolation of nuclear and cytosolic fractions was carried out by lysing the cells for 10 min on ice in buffer A (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCL, 0.5 mM DTT, 0.5% NP40, pH7.9) followed by centrifugation at 3000 r.p.m. for 10 min. The supernatants were retained as the cytosolic fractions, and the pellets were subjected to further lysis in buffer B (5 mM HEPES, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 26% glycerol (v/v), pH 7.9). NaCl was added to a final concentration of 300 mM, and the samples were homogenized using a Dounce homogenizer (VWR, Dublin, Ireland). After a 30-min incubation on ice, the lysates were centrifuged for 20 min at 24 000 g, and the resulting supernatants collected as the nuclear fractions.
Antibodies
Mouse monoclonal (Western): FLIP (NF6; Alexis, San Diego, CA, USA), Caspase 8 (12F5; Alexis) and PARP (eBioscience, San Diego, CA, USA), β-Actin and Flag-HRP (Sigma, Dorset, UK), Ku70, ITCH (both BD Transduction Laboratories, Oxfordshire, UK) and Noxa (Abcam, Cambridge, UK). Rabbit polyclonal (Western and IP): Caspase 9, caspase 3, acetylated lysine, acetylated-α-tubulin, XIAP, Bcl-2, Bcl-XL, Mcl-1, Bax, Bak, Bid, Bim, PUMA (Cell Signaling Technology, Beverly, MA, USA), c-FLIP (H202), Ku70 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and Hyperacetylated Histone H4 (Millipore, Watford, UK). Horseradish peroxidase-conjugated sheep anti-mouse and sheep anti-rabbit secondary antibodies were used as appropriate (Amersham, Buckinghamshire, UK).
Yeast-2-hybrid
The Matchmaker GAL4-based yeast-two-hybrid system was used (Clontech, Saint Germain en Laye, France). Human FLIP(L) was used as the bait protein and expressed as a fusion to the GAL4 DNA-binding domain in pGBKT7 vector. A cDNA library generated from HCT116 cells was cloned into pGADT7 prey vector. The yeast-two-hybrid screen for FLIP(L)-interacting proteins was carried out according to the manufacturer's instructions.
Immunoprecipitation (IP) reactions
Protein lysates were prepared using CHAPS (10 mM HEPES, 150 mM NaCl, 1% CHAPS, pH 7.4) or NP-40 (0.2% NP-40, 20 mM Tris-HCL (pH 7.4), 150 mM NaCl, 10% glycerol) buffers. Lysates (0.5–2 mg) were pre-cleared overnight with sheep anti-rabbit IgG Dynalbeads (Invitrogen, Paisley, UK). Antibodies (1–2 μg) were conjugated with sheep anti-rabbit IgG Dynal beads for at least 1 h and then washed before incubation with pre-cleared lysates for at least 4 h. After several washes, Dynal beads were resuspended in Laemmli loading buffer and heated at 95 °C for 5 min before immunoblot analysis. Ubiquitin IP experiments were carried out using the Ubiquitin Enrichment kit (Thermo Scientific, Surrey, UK) according to the manufacturer's instructions.
Recombinant protein purification and GST pull-downs
GST and GST-tagged FLIP(S) were expressed in IPTG-stimulated BL21 bacteria. Bacterial cell lysates were purified using Glutathione-Sepharose beads (GE Healthcare Bio-Sciences AB, Uppsala, Sweden) and dialysed using Slide-A-Lyzer Dialysis Cassettes (Thermo Scientific). For pull-downs, GST-tagged proteins (10 μg) were conjugated to Glutathione-Sepharose beads and incubated with pre-cleared protein lysates (500 μg) for 2 h at 4 °C with constant mixing. Beads were washed several times and resuspended in Laemmli loading buffer before western blot analysis.
FLIP peptide array analysis
FLIP peptide arrays were generated as previously described9 using the Genbank sequence U97074.1. The arrays were blocked in 5% milk/PBS/0.5% Tween-20 for 1 h and then overlaid with lysates prepared from the cells transfected with Flag-tagged Ku70 or EV. Following several washes (PBS/0.5% Tween-20), bound Flag-tagged Ku70 was detected by immunoblotting.
Caspase ‘trap’ assay
This assay was performed as previously described.17
Flow cytometry
Apoptosis was determined using propidium iodide (PI) staining to evaluate the percentage of cells with DNA content <2N as previously described.3 For annexin V/PI analysis, cells were harvested and analysed according to the manufacturer's instructions (BD Biosciences, Oxford, UK).
siRNA transfections
The non-silencing control (SC) siRNA, FLIP-, caspase 8-, caspase 9, DR4- and DR5-targeted siRNAs were obtained from Dharmacon (Chicago, IL, USA) and were the same as those previously described.3, 40 siRNAs targeting Ku70 and HDAC6 were obtained from Qiagen (West Sussex, UK). siRNA transfections were carried out using OligofectAMINE (Invitrogen).
Quantitative PCR (Q-PCR)
RNA was isolated using the RNA STAT-60 reagent (Biogenesis, Poole, UK) and reverse transcribed using Moloney murine leukemia virus-based reverse transcriptase kit (Invitrogen). Q-PCR analysis of the FLIP gene expression was performed using the SYBR Green method or TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA, USA).
Animal model experiments
Female BALB/c nude mice were maintained as previously described.3 All experiments were carried out in accordance with the Animals (Scientific Procedures) Act, 1986 under a Project Licence (PPL 2590b) approved by the Department of Health, Social Services and Public Safety, Northern Ireland. Mice were implanted sub-cutaneously on each flank with 2 × 106 HCT116 or FL17 cells using Matrigel (BD, Oxford, UK). After the tumours reached ∼100 mm3, mice were randomised to receive vehicle control (10% DMSO/45% PEG400) or 100 mg/kg Vorinostat delivered IP on a 5-day on/2-day off schedule for 2 weeks. Tumour volumes were determined as previously described.3 To extract protein, xenografts were homogenized in RIPA buffer.
Caspase activity assay
Caspase-8 or caspase-3/7-GLO reagents (25 μl) (Promega, Southampton, UK) were incubated with 1–10 μg of protein lysate diluted in cell culture medium in a total volume of 50 μl for 1–2 h at room temperature. Luciferase activity was then determined using a luminometer.
Statistical analysis
Student's t-test was used for the statistical analysis; *denotes P<0.05;. **denotes P<0.01; ***denotes P<0.001; and ns denotes not significant compared with control.
Abbreviations
- CBP:
-
CREB-binding protein
- CRC:
-
colorectal cancer
- DED:
-
death effector domain
- DISC:
-
death-inducing signaling complex
- DR4 and DR5:
-
death receptor 4 and 5
- DUB:
-
de-ubiquitinating enzyme
- FLIP:
-
Fas-associated death domain (FADD)-like interleukin 1b-converting enzyme (FLICE)-inhibitory protein
- 5-FU:
-
5-fluorouracil
- HAT:
-
histone acetyl transferase
- HDAC:
-
histone deacetylase
- HSP90:
-
heat shock protein 90
- NHEJ:
-
non-homologous end-joining
- PARP:
-
poly(ADP-ribose) polymerase
- PCAF:
-
P300/CBP-associated factor
- Q-PCR:
-
quantitative PCR
- SAHA:
-
suberoylanilide hydroxamic acid
- TRAIL:
-
TNF-related apoptosis-inducing ligand
- UPS:
-
ubiquitin proteasome system
- XIAP:
-
X-linked inhibitor of apoptosis
References
Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V et al. Inhibition of death receptor signals by cellular FLIP. Nature 1997; 388: 190–195.
Scaffidi C, Schmitz I, Krammer PH, Peter ME . The role of c-FLIP in modulation of CD95-induced apoptosis. J Biol Chem 1999; 274: 1541–1548.
Wilson TR, McLaughlin KM, McEwan M, Sakai H, Rogers KM, Redmond KM et al. c-FLIP: a key regulator of colorectal cancer cell death. Cancer Res 2007; 67: 5754–5762.
McLornan DP, Barrett HL, Cummins R, McDermott U, McDowell C, Conlon SJ et al. Prognostic significance of TRAIL signaling molecules in stage II and III colorectal cancer. Clin Cancer Res 2010; 16: 3442–3451.
Featherstone C, Jackson SP . Ku, a DNA repair protein with multiple cellular functions? Mutat Res 1999; 434: 3–15.
Cohen HY, Lavu S, Bitterman KJ, Hekking B, Imahiyerobo TA, Miller C et al. Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis. Mol Cell 2004; 13: 627–638.
Chen CS, Wang YC, Yang HC, Huang PH, Kulp SK, Yang CC et al. Histone deacetylase inhibitors sensitize prostate cancer cells to agents that produce DNA double-strand breaks by targeting Ku70 acetylation. Cancer Res 2007; 67: 5318–5327.
Amsel AD, Rathaus M, Kronman N, Cohen HY . Regulation of the proapoptotic factor Bax by Ku70-dependent deubiquitylation. Proc Natl Acad Sci USA 2008; 105: 5117–5122.
Kiely PA, Baillie GS, Lynch MJ, Houslay MD, O’Connor R . Tyrosine 302 in RACK1 is essential for insulin-like growth factor-I-mediated competitive binding of PP2A and beta1 integrin and for tumor cell proliferation and migration. J Biol Chem 2008; 283: 22952–22961.
Yang JK, Wang L, Zheng L, Wan F, Ahmed M, Lenardo MJ et al. Crystal structure of MC159 reveals molecular mechanism of DISC assembly and FLIP inhibition. Mol Cell 2005; 20: 939–949.
Ueffing N, Keil E, Freund C, Kuhne R, Schulze-Osthoff K, Schmitz I . Mutational analyses of c-FLIPR, the only murine short FLIP isoform, reveal requirements for DISC recruitment. Cell Death Differ 2008; 15: 773–782.
Guo F, Sigua C, Tao J, Bali P, George P, Li Y et al. Cotreatment with histone deacetylase inhibitor LAQ824 enhances Apo-2 L/tumor necrosis factor-related apoptosis inducing ligand-induced death inducing signaling complex activity and apoptosis of human acute leukemia cells. Cancer Res 2004; 64: 2580–2589.
Watanabe K, Okamoto K, Yonehara S . Sensitization of osteosarcoma cells to death receptor-mediated apoptosis by HDAC inhibitors through downregulation of cellular FLIP. Cell Death Differ 2005; 12: 10–18.
Wood TE, Dalili S, Simpson CD, Sukhai MA, Hurren R, Anyiwe K et al. Selective inhibition of histone deacetylases sensitizes malignant cells to death receptor ligands. Mol Cancer Ther 2010; 9: 246–256.
Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol 2010; 6: 238–243.
Schimmer AD, Thomas MP, Hurren R, Gronda M, Pellecchia M, Pond GR et al. Identification of small molecules that sensitize resistant tumor cells to tumor necrosis factor-family death receptors. Cancer Res 2006; 66: 2367–2375.
Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR . In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol 2006; 8: 72–77.
Yamaguchi H, Woods NT, Piluso LG, Lee HH, Chen J, Bhalla KN et al. p53 acetylation is crucial for its transcription-independent proapoptotic functions. J Biol Chem 2009; 284: 11171–11183.
Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL . Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA 2003; 100: 4389–4394.
Longley DB, Wilson TR, McEwan M, Allen WL, McDermott U, Galligan L et al. c-FLIP inhibits chemotherapy-induced colorectal cancer cell death. Oncogene 2006; 25: 838–848.
Jeggo PA . Identification of genes involved in repair of DNA double-strand breaks in mammalian cells. Radiat Res 1998; 150: S80–S91.
Wang J, Dong X, Myung K, Hendrickson EA, Reeves WH . Identification of two domains of the p70 Ku protein mediating dimerization with p80 and DNA binding. J Biol Chem 1998; 273: 842–848.
Frew AJ, Lindemann RK, Martin BP, Clarke CJ, Sharkey J, Anthony DA et al. Combination therapy of established cancer using a histone deacetylase inhibitor and a TRAIL receptor agonist. Proc Natl Acad Sci USA 2008; 105: 11317–11322.
Kauh J, Fan S, Xia M, Yue P, Yang L, Khuri FR et al. c-FLIP degradation mediates sensitization of pancreatic cancer cells to TRAIL-induced apoptosis by the histone deacetylase inhibitor LBH589. PLoS One 2010; 5: e10376.
Subramanian C, Jarzembowski JA, Opipari Jr AW, Castle VP, Kwok RP . HDAC6 deacetylates Ku70 and regulates Ku70-Bax binding in neuroblastoma. Neoplasia 2010; 13: 726–734.
Boyault C, Sadoul K, Pabion M, Khochbin S . HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007; 26: 5468–5476.
Schuchmann M, Schulze-Bergkamen H, Fleischer B, Schattenberg JM, Siebler J, Weinmann A et al. Histone deacetylase inhibition by valproic acid down-regulates c-FLIP/CASH and sensitizes hepatoma cells towards CD95- and TRAIL receptor-mediated apoptosis and chemotherapy. Oncol Rep 2006; 15: 227–230.
Pathil A, Armeanu S, Venturelli S, Mascagni P, Weiss TS, Gregor M et al. HDAC inhibitor treatment of hepatoma cells induces both TRAIL-independent apoptosis and restoration of sensitivity to TRAIL. Hepatology 2006; 43: 425–434.
Minucci S, Pelicci PG . Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer 2006; 6: 38–51.
Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol 2005; 23: 3923–3931.
Blumenschein Jr GR, Kies MS, Papadimitrakopoulou VA, Lu C, Kumar AJ, Ricker JL et al. Phase II trial of the histone deacetylase inhibitor vorinostat (Zolinza, suberoylanilide hydroxamic acid, SAHA) in patients with recurrent and/or metastatic head and neck cancer. Invest New Drugs 2008; 26: 81–87.
Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res 2003; 9: 3578–3588.
Kavanaugh SM, White LA, Kolesar JM . Vorinostat: A novel therapy for the treatment of cutaneous T-cell lymphoma. Am J Health Syst Pharm 2010; 67: 793–797.
Fakih MG, Fetterly G, Egorin MJ, Muindi JR, Espinoza-Delgado I, Zwiebel JA et al. A phase I, pharmacokinetic, and pharmacodynamic study of two schedules of vorinostat in combination with 5-fluorouracil and leucovorin in patients with refractory solid tumors. Clin Cancer Res 2010; 16: 3786–3794.
Fakih MG, Pendyala L, Fetterly G, Toth K, Zwiebel JA, Espinoza-Delgado I et al. A phase I, pharmacokinetic and pharmacodynamic study on vorinostat in combination with 5-fluorouracil, leucovorin, and oxaliplatin in patients with refractory colorectal cancer. Clin Cancer Res 2009; 15: 3189–3195.
Wilson PM, El-Khoueiry A, Iqbal S, Fazzone W, LaBonte MJ, Groshen S et al. A phase I/II trial of vorinostat in combination with 5-fluorouracil in patients with metastatic colorectal cancer who previously failed 5-FU-based chemotherapy. Cancer Chemother Pharmacol 2010; 65: 979–988.
Itoh Y, Suzuki T, Kouketsu A, Suzuki N, Maeda S, Yoshida M et al. Design, synthesis, structure—selectivity relationship, and effect on human cancer cells of a novel series of histone deacetylase 6-selective inhibitors. J Med Chem 2007; 50: 5425–5438.
Gediya LK, Belosay A, Khandelwal A, Purushottamachar P, Njar VC . Improved synthesis of histone deacetylase inhibitors (HDIs) (MS-275 and CI-994) and inhibitory effects of HDIs alone or in combination with RAMBAs or retinoids on growth of human LNCaP prostate cancer cells and tumor xenografts. Bioorg Med Chem 2008; 16: 3352–3360.
McDermott U, Longley DB, Galligan L, Allen W, Wilson T, Johnston PG . Effect of p53 status and STAT1 on chemotherapy-induced, Fas-mediated apoptosis in colorectal cancer. Cancer Res 2005; 65: 8951–8960.
Wilson TR, McEwan M, McLaughlin K, Le Clorennec C, Allen WL, Fennell DA et al. Combined inhibition of FLIP and XIAP induces Bax-independent apoptosis in type II colorectal cancer cells. Oncogene 2009; 28: 63–72.
Acknowledgements
This work was supported by a grant from Cancer Research UK. EK was supported by a studentship from the Department for Employment and Learning, NI.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
PGJ is a consultant for Merck. The other authors declare no conflict of interest.
Additional information
Edited by E Gottlieb
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Kerr, E., Holohan, C., McLaughlin, K. et al. Identification of an acetylation-dependant Ku70/FLIP complex that regulates FLIP expression and HDAC inhibitor-induced apoptosis. Cell Death Differ 19, 1317–1327 (2012). https://doi.org/10.1038/cdd.2012.8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2012.8
Keywords
This article is cited by
-
UBE4B interacts with the ITCH E3 ubiquitin ligase to induce Ku70 and c-FLIPL polyubiquitination and enhanced neuroblastoma apoptosis
Cell Death & Disease (2023)
-
HDAC6: A unique HDAC family member as a cancer target
Cellular Oncology (2022)
-
Glucocorticoid modulatory element-binding protein 1 (GMEB1) interacts with the de-ubiquitinase USP40 to stabilize CFLARL and inhibit apoptosis in human non-small cell lung cancer cells
Journal of Experimental & Clinical Cancer Research (2019)
-
The arginine methyltransferase PRMT5 and PRMT1 distinctly regulate the degradation of anti-apoptotic protein CFLARL in human lung cancer cells
Journal of Experimental & Clinical Cancer Research (2019)
-
Droxinostat sensitizes human colon cancer cells to apoptotic cell death via induction of oxidative stress
Cellular & Molecular Biology Letters (2018)
